- Lymphangioleiomyomatose
-
Klassifikation nach ICD-10 D48 Neubildung unsicheren oder unbekannten Verhaltens an sonstigen und nicht näher bezeichneten Lokalisationen D48.1 Bindegewebe und andere Weichteilgewebe
- Lymphangioleiomyomatose[1]ICD-10 online (WHO-Version 2011) 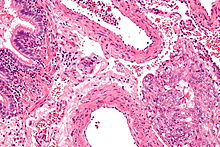 Mikrofoto von Lymphangioleiomyomatose. HE-Färbung.
Mikrofoto von Lymphangioleiomyomatose. HE-Färbung.
Lymphangioleiomyomatose (LAM) ist eine sehr seltene Erkrankung der Lunge, die nahezu ausschließlich bei Frauen auftritt. Sie ist meistens fortschreitend, führt zu chronischem Sauerstoffmangel und ist schließlich lebensbedrohend.
Die Krankheit gibt es in zwei verschiedenen Formen: einmal als sogenannte sporadische LAM, die nicht vererbt werden kann, und andererseits als LAM, die im Zusammenhang mit der Erkrankung Tuberöse Sklerose auftritt und vererbbar ist. Die ersten Anzeichen der Erkrankung, wie Luftnot bei Belastung oder Husten und Schmerzen im Brustkorb, treten meistens schon im Alter zwischen 25 und 30 Jahren auf. Weil LAM so selten ist und meist schleichend beginnt, bleiben die Beschwerden oft ohne korrekte Diagnose oder werden als Asthma oder Lungenemphysem fehldiagnostiziert. Die eindeutige Diagnose ist mittels Computertomographie oder durch eine Lungenbiopsie möglich. Der CT-radiologische Befund[2] ist sehr typisch und erlaubt in aller Regel die Diagnosestellung. Es wird geschätzt, dass es in Deutschland rund 200 Patientinnen mit LAM gibt.
Bis heute ist die Ursache von LAM nicht vollständig aufgeklärt. Vermutet wird eine genetische Störung, die zu gefährlichen Veränderungen der Lungen und der Nieren führt. In den Lungen zerstört ein unkontrolliertes Wachstum der sogenannten glatten Muskelzellen zunehmend das gesunde Lungengewebe und schränkt damit die Sauerstoffaufnahme des Körpers immer mehr ein. Diese Verwachsungen sind chirurgisch teilweise entfernbar. Dies steigert die Lebensqualität, die Zysten sind jedoch rezidiv.
Der makroskopische Befund[3] ähnelt einem schweren Emphysem mit verbreiterten Alveolarsepten. Dadurch wird das Atmen für Patienten mit LAM immer schwerer und sie sind körperlich wenig belastbar. LAM verursacht bei einem Teil der Betroffenen auch Angiomyolipome der Nieren oder fibrotische Gewebeveränderungen im Bauchraum und vergrößerte Lymphknoten. Bei der Hälfte aller Betroffenen kommt es im Verlauf der Erkrankung zu schweren Symptomen wie zum Pneumothorax (Kollaps der Lunge), und bei circa 15 % der Frauen zum Chylothorax (Ansammlung von Lymphflüssigkeit im Pleuraspalt).
Obwohl in den USA vor einigen Jahren intensive Forschungen über LAM begonnen wurden, ist noch keine Therapie bekannt, die wissenschaftlich nachweisbar wirkt. Die bisher gängige Therapie mit Medroxy-Progesteron scheint nur bei einem Teil der Patienten bedingt zu wirken. Viele Betroffene und Ärzte stehen der Krankheit völlig hilflos gegenüber und sehen im fortgeschrittenen Krankheitsstadium als einzige Chance die Lungentransplantation, die jedoch nur für jüngere Patienten in gutem Allgemeinzustand infrage kommt. Die Prognose nach einer Lungentransplantation ist gut. Rezidive sind bislang nicht beschrieben. Neue Forschungen in den USA mit dem Medikament Sirolimus (Rapamycin) lassen für die Zukunft auf eine wirksame Therapie hoffen.
Bislang konnte die Krankheit nur bei einem männlichen Patienten eindeutig nachgewiesen werden.[4]
Einzelnachweise
- ↑ med-kolleg.de
- ↑ Gerald F. Abbott, Melissa L. Rosado-de-Christenson, Aletta Ann Frazier, Teri J. Franks, Robert D. Pugatch, Jeffrey R. Galvin. Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation. RadioGraphics 2005;25:803-828 doi:10.1148/rg.253055006
- ↑ Abbildung aus RadioGraphics
- ↑ A.J.R.C.C.M.: Pulmonary Lymphangioleiomyomatosis in a Man

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in der Pneumologie
- Erbkrankheit
Wikimedia Foundation.