- Morbus Osler
-
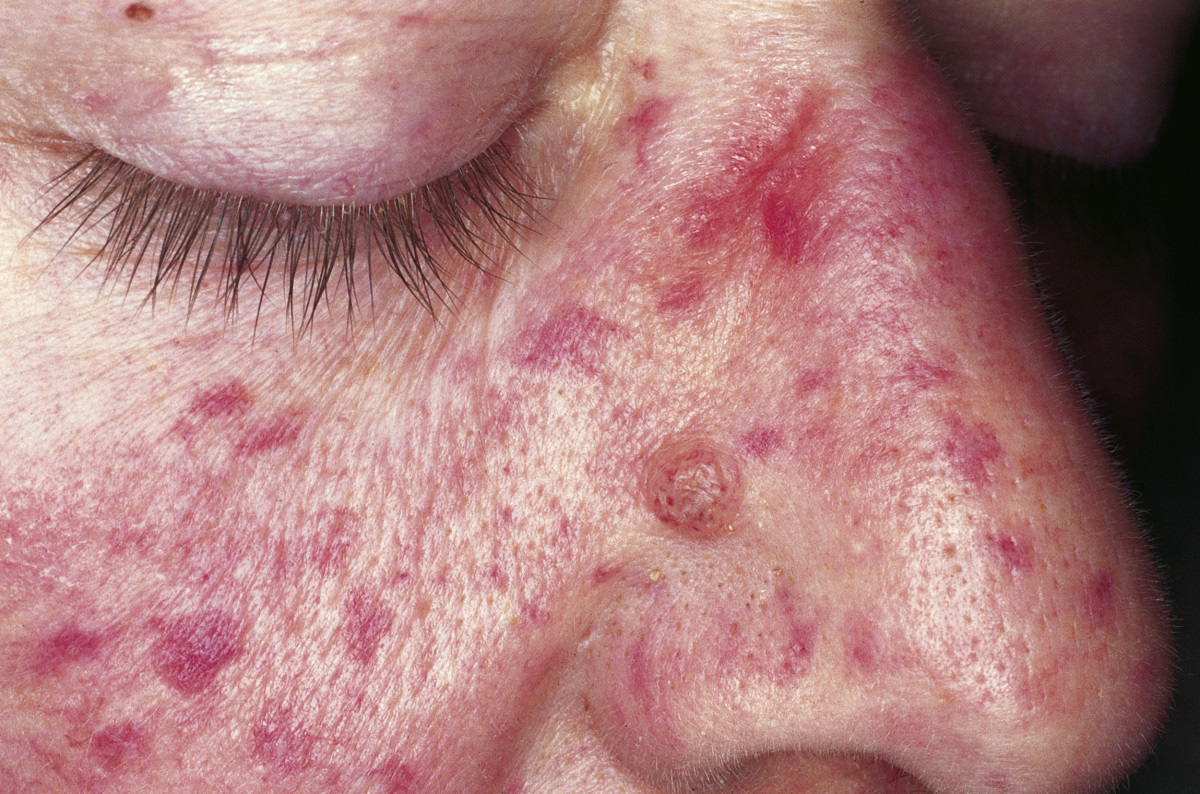
Klassifikation nach ICD-10 I78.0 Hereditäre hämorrhagische Teleangiektasie ICD-10 online (WHO-Version 2011) 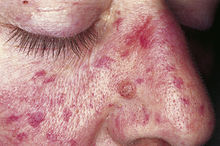 Patient mit Morbus Osler[1]
Patient mit Morbus Osler[1]
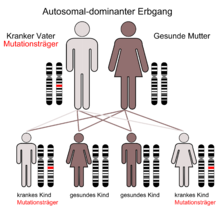 Der autosomal-dominante Erbgang
Der autosomal-dominante ErbgangMorbus Osler oder Osler-Syndrom (nach William Osler), auch Morbus Osler-Weber-Rendu, Morbus Osler-Rendu-Weber oder Hereditäre hämorrhagische Teleangiektasie (HHT) ist eine autosomal-dominant vererbte Erkrankung, bei der es zu einer krankhaften Erweiterung von Blutgefäßen kommt. Diese sogenannten Teleangiektasien können überall auftreten, finden sich jedoch besonders in Nase (daher auch das Leitsymptom Nasenbluten), Mund, Gesicht und den Schleimhäuten des Magen-Darm-Traktes. Da die Gefäßerweiterungen sehr verletzlich sind, kann es leicht zu Einrissen und somit zur Blutung kommen.
Unter anderem weiten sich kleinste Gefäße von Haut und Schleimhaut und sind anschließend als stecknadelkopf- bis reiskorngroße rote Flecken zu sehen. Besondere Bedeutung haben diese Teleangiektasien im Magen-Darm-Trakt, weil sie dort Ursache für häufig wiederkehrende (rezidivierende) Blutungen sein können. Es können jedoch auch bedeutend größere Gefäßerweiterungen auftreten. Diese entstehen besonders in der Lunge, dem Gehirn und der Leber. Die Veränderungen machen sich oft lange Zeit nicht bemerkbar, können jedoch z. B. durch Blutungen plötzlich sehr bedrohlich werden (s. u.). Die ersten Anzeichen der Erkrankung zeigen sich meist in der Pubertät mit Nasenbluten, bei wenigen Patienten jedoch auch ohne Nasenbluten und zum Teil viel später.
In Deutschland leiden rund 35.000 Menschen an M. Osler.
Inhaltsverzeichnis
Ursachen
Die Erkrankung wird autosomal-dominant vererbt. Dies bedeutet, dass bei einem Elternpaar, bei dem ein Partner Morbus Osler-Patient ist, im Durchschnitt die Hälfte der Kinder unabhängig vom Geschlecht betroffen sind.
Es gibt mindestens drei Gene, die im veränderten Zustand (Mutation) zum Krankheitsbild des Morbus Osler führen können. Zwei dieser Gene sind heute bekannt und können bei Patienten und ihren Familienangehörigen untersucht werden (Endoglin, ein TGFβ1-Rezeptor, auf Chromosom 9q und ALK-1 = activin receptor like kinase 1 auf Chromosom 12q)*[2]. Möglicherweise gibt es auch Patienten, die genetisch Morbus-Osler-Patienten sind, die jedoch nie Anzeichen der Erkrankung zeigen.
Diagnostik
Die Diagnose wird überwiegend klinisch gestellt, genetische Untersuchungen können jedoch entscheidend dazu beitragen.
Kriterien zur klinischen Diagnostik des Morbus Osler (HHT) (sog. Curaçao-Kriterien), erstellt vom medizinischen und wissenschaftlichen Beratungsgremium der amerikanischen Selbsthilfegruppe:
- Epistaxis - Nasenbluten (spontan und wiederholt)
- Teleangiektasien - Typische kleine Gefäßmissbildungen, mehrfach und an charakteristischen Stellen (Lippen, Mundhöhle, Finger, Nase)
- Viszerale Manifestationen - Beteiligung innerer Organe, besonders von Lunge, Leber, Hirn und Magen-Darm-Trakt (s. Text)
- Positive Familienanamnese - Wenigstens ein Verwandter ersten Grades, der nach diesen Kriterien betroffen ist.
Der Morbus Osler gilt als gesichert, wenn wenigstens drei dieser vier Kriterien erfüllt sind. Bei zwei erfüllten Kriterien geht man von einem Verdachtsfall aus. Auch wenn bei nur einem erfüllten Kriterium ein Morbus Osler unwahrscheinlich ist, ist dieser trotzdem möglich, z. B. betroffene Kinder, bei denen häufig nur der vierte Punkt erfüllt ist, während sich die anderen erst im Laufe des Lebens einstellen können. Hier ist häufig die genetische Diagnostik hilfreich.
Symptome und Behandlung
Nasenbluten
90% aller Menschen mit Morbus Osler haben Nasenbluten, das meist bereits in der Pubertät auftritt und Ausmaße annehmen kann, die zu einer Einschränkung der Lebensqualität bis hin zur Arbeitsunfähigkeit führen können. Bei vielen Patienten kommt es zur Blutarmut (Anämie). Es kann erforderlich sein, Eisen oder Blut zuzuführen. Zur Vorbeugung werden Nasensalben verwendet, im Blutungsfall eine Nasentamponade. Operative Therapien sind die Laserlichtbehandlung der Gefäßerweiterung oder die Transplantation, bei der die erkrankte Nasenschleimhaut durch Haut aus anderen Gesichtsregionen (zum Beispiel von hinter dem Ohr) ersetzt wird. Eine Heilung auf Dauer ist bislang noch nicht möglich.
Lunge
Ungefähr 5 bis 30 % aller Morbus-Osler-Patienten haben große Gefäßerweiterungen in den Lungen, sogenannte pulmonale arteriovenöse Malformationen (PAVM). Durch diese großen Gefäßkurzschlüsse können auch Gerinnsel und Bakterien passieren und so zu Schlaganfällen und Hirneiterungen (Abszessen) führen. Bei ärztlichen Eingriffen, insbesondere bei Zahnbehandlungen, kann es zur Einschwemmung von Bakterien in das Blut kommen. Deshalb wird empfohlen, dass alle Patienten, bei denen eine PAVM vorliegen könnte, vorher Antibiotika erhalten. Besonders während der Schwangerschaft können die abnormen Gefäße der Lunge deutlich an Größe zunehmen. Wenn der Kurzschluss zu groß wird, kann es zur Überlastung des Kreislaufs kommen. Lungenblutungen treten eher selten auf, können jedoch lebensbedrohlich sein. Bei Verdacht auf eine PAVM wird eine Computertomographie (CT) oder Kernspintomographie des Brustkorbs, eine Blutgasanalyse oder geeignete Ultraschalluntersuchungen durchgeführt.
Wenn erforderlich, lassen sich die erweiterten Gefäße durch das Einbringen von Metallspiralen oder kleinen Ballons verschließen (Embolisation). Selten sind jedoch auch große Operationen mit Öffnung des Brustkorbs angezeigt.
Gehirn
Wie oben erläutert können bei Lungengefäßerweiterungen (PAVM) Gerinnsel und Bakterien die Lunge passieren und zu Schlaganfällen oder Hirnabszessen führen. Zusätzlich können auch cerebrale vaskuläre Malformationen (CVM, cerebral „zum Hirn gehörend“, vaskulär steht für Gefäß) auftreten. Auch bei den CVM können Blutungen auftreten, es muss jedoch nicht jede CVM behandelt werden. Es muss abgewogen werden, wie groß das Blutungsrisiko im Vergleich zum Behandlungsrisiko ist. Die Gefäßmissbildungen können durch Einbringen von Material über einen Katheter verschlossen (Embolisation) oder nach Öffnung der Schädeldecke operativ entfernt werden. Man schätzt die Häufigkeit der CVM auf 5 bis 20%, deshalb empfiehlt die amerikanische Selbsthilfeorganisation den Morbus-Osler-Patienten, dass bei allen Patienten nach dem 12. Lebensjahr eine spezielle Kernspintomographie des Kopfes durchgeführt werden sollte. Bei Verdachtsmomenten (z.B. Kopfschmerzen oder Lähmungen) kann dies jedoch bereits früher sinnvoll sein.
Magen-Darm-Trakt
Besonders ab dem 40. Lebensjahr kann es zu Magen- und Darmblutungen kommen. Diese Blutungen können geringgradig, jedoch auch sehr heftig sein. Der Stuhl kann bei starken Blutungen teerähnlich dunkel aussehen und „faul“ riechen oder mit rotem Stuhl durchmengt sein. Bei leichten Blutungen können diese Zeichen jedoch unbemerkt bleiben. Manchmal bringt erst die Abklärung einer Blutarmut, die vom Patienten meist als allgemeine Schwäche und Müdigkeit verspürt wird, den Arzt auf die richtige Spur. Zur Abklärung wird dann häufig eine Magen-Darm-Spiegelung durchgeführt. Finden sich dort wenige und geeignete Gefäßerweiterungen, so kann eine Behandlung mittels Laser, Unterspritzung oder elektrischer Verödung oft im gleichen Eingriff durchgeführt werden. Liegen viele Blutungsquellen vor und kommt es zu wiederholten Blutungen, so kann eine Therapie mit weiblichen Hormonen versucht werden. Aufgrund der möglichen Nebenwirkungen (unter anderem auf das Herz-Kreislauf-System und Brustbildung bei Männern) muss ein sorgfältiges Abwägen der Vor- und Nachteile erfolgen.
Leber
Auch in der Leber können Gefäßkurzschlüsse, sogenannte "Shunts", auftreten. Diese können zu einer Überlastung des Herzens führen, was von den Betroffenen häufig als Abgeschlagenheit und mangelnde körperliche Belastbarkeit, ähnlich wie bei der Blutarmut, empfunden wird. Die medikamentöse Verbesserung der Herzfunktion wird häufig als erster Schritt der Behandlung versucht. Es gibt eine Reihe eingreifender Behandlungen, insbesondere die Embolisation (s. o.) und die Lebertransplantation. Da die Nebenwirkungen gravierend sein können, ist hier ähnlich wie bei den CVM ein sorgfältiges Abwägen der Vor- und Nachteile erforderlich.
Einzelnachweise
- ↑ M. Sand, D. Sand, C. Thrandorf, V. Paech, P. Altmeyer, F. G. Bechara: Cutaneous lesions of the nose. In: Head & face medicine Band 6, 2010, S. 7, ISSN 1746-160X. doi:10.1186/1746-160X-6-7. PMID 20525327. PMC 290354. (Review).
- ↑ Nguyen NQ: Hereditary hemorrhagic telangiectasia. Dermatol Online J. 2005 Dec 30;11(4):19. PMID 16403391
Weblinks

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in der Hals-Nasen-Ohren-Heilkunde
- Krankheitsbild in der Gastroenterologie
- Krankheitsbild in der Angiologie
- Erbkrankheit
- Angiodysplasie
Wikimedia Foundation.