- Papillärer Tumor der Pinealisregion
-
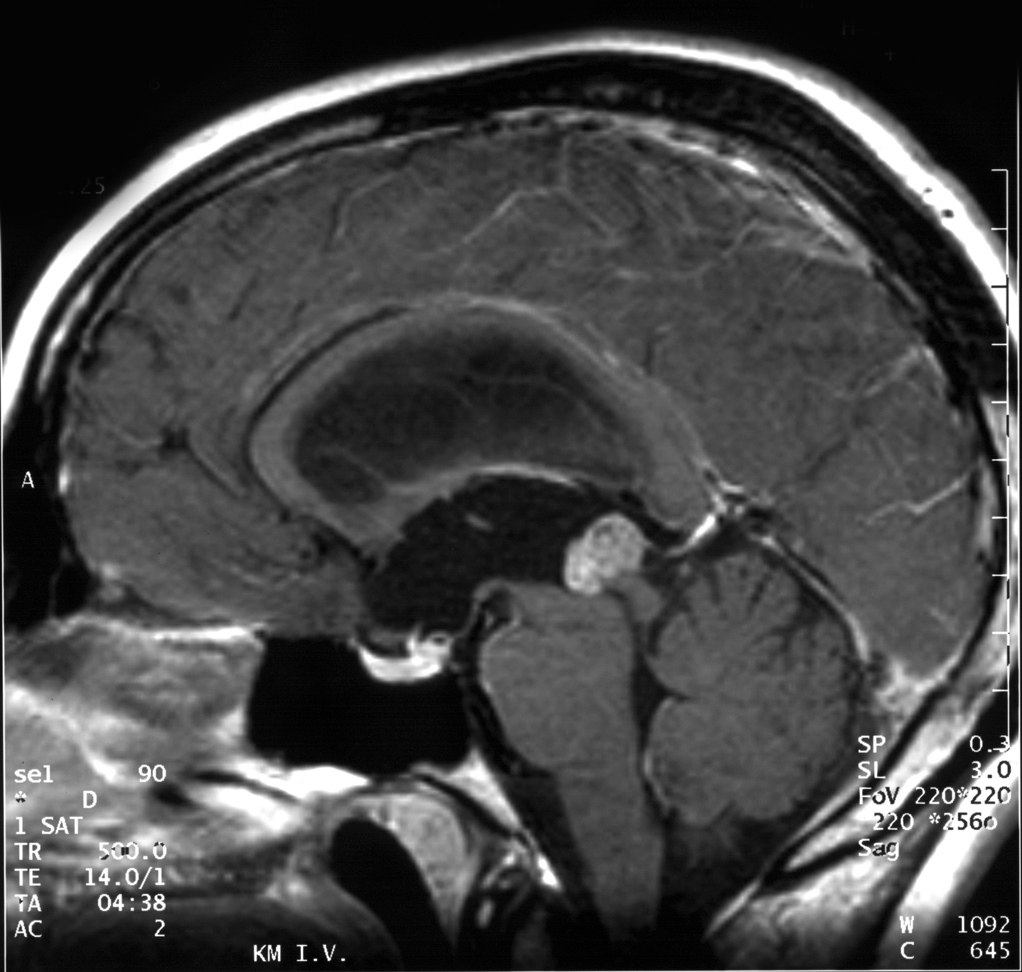
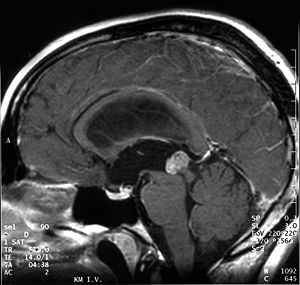 Kernspintomographie. Papillärer Tumor der Pinealisregion bei einem 18-jährigen Patienten (sagittale T1 gewichtete Aufnahme nach Kontrastmittel)
Kernspintomographie. Papillärer Tumor der Pinealisregion bei einem 18-jährigen Patienten (sagittale T1 gewichtete Aufnahme nach Kontrastmittel)
Klassifikation nach ICD-10 D43 Neubildung unsicheren oder unbekannten Verhaltens des Gehirns und des Zentralnervensystems D43.0 Gehirn, supratentoriell ICD-10 online (WHO-Version 2011) 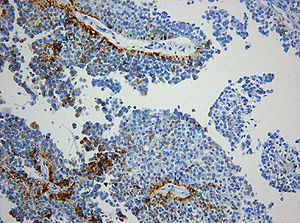 Immunhistochemie. Expression von Zytokeratin (Klon KL1) in einem papillären Tumor der Pinealisregion. Immunhistochemie. Originalvergrößerung 1:200
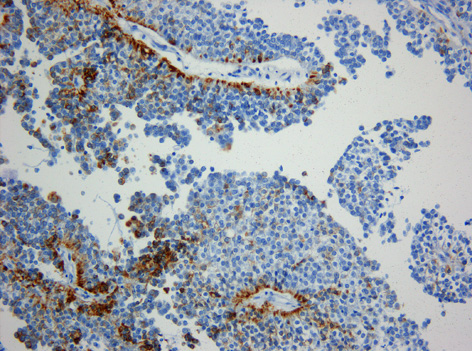
Immunhistochemie. Expression von Zytokeratin (Klon KL1) in einem papillären Tumor der Pinealisregion. Immunhistochemie. Originalvergrößerung 1:200 Immunhistochemie. Fehlende Expression des Epithelialen-Membran-Antigens (EMA) in einem papillären Tumor der Pinealisregion. Originalvergrößerung 1:200
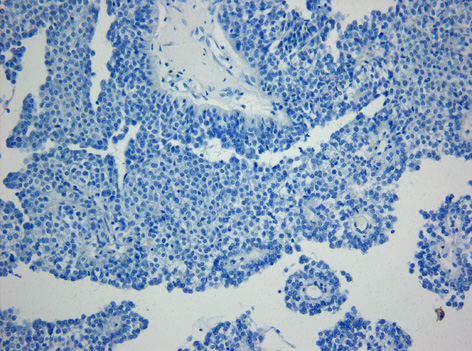
Immunhistochemie. Fehlende Expression des Epithelialen-Membran-Antigens (EMA) in einem papillären Tumor der Pinealisregion. Originalvergrößerung 1:200Der papilläre Tumor der Pinealisregion, kurz auch PTPR, ist ein seltener Hirntumor, der vermutlich von spezialisierten Ependymzellen des Subcommissuralorgans ausgeht. Papilläre Tumoren der Pinealisregion sind an der Rückwand des dritten Ventrikels gelegen und stehen in enger räumlicher Beziehung zur Zirbeldrüse (Glandula Pinealis). Nach der Erstbeschreibung im Jahre 2003 [1] sind über 40 Fälle publiziert worden. In der ganz überwiegenden Mehrzahl sind Kinder und junge Erwachsene betroffen.[2] Ein Zusammenhang mit einer erblichen Erkrankung ist nicht bekannt.
Inhaltsverzeichnis
Klinische Manifestationen
Aufgrund ihrer typischen Lage behindern papilläre Tumoren der Pinealisregion häufig den Fluss der Cerebrospinalflüssigkeit, was zu einer Erhöhung des Hirndrucks führen kann. Kopfschmerz, Übelkeit und Erbrechen sind unspezifische Symptome. Durch Druck des Tumors auf die Vierhügelplatte treten nicht selten Störungen der Okulomotorik mit Doppelbildern auf (Parinaud-Syndrom). In der Kernspintomographie stellen sich papilläre Tumoren der Pinealisregion als kontrastmittelanreichernde Raumforderungen dar.
Pathologie
Histologisch handelt es sich um einen neuroektodermalen Tumor, der mit einem papillären Aufbau ein epithel-ähnliches Aussehen hat. Die Tumorzellen sitzen häufig mit breiten Fortsätzen den Blutgefäßen auf, so dass der Eindruck plumper Pseudorosetten entsteht. Auch ependymale Rosetten wie bei Ependymomen können vorkommen. Die mitotische Aktivität der Tumorzellen ist meist moderat. Nekrosen sind häufig und können zur Fehldiagnose eines Plexuskarzinoms verleiten. In der Vergangenheit sind papilläre Tumoren der Pinealisregion häufig auch als Plexuspapillom oder papilläres Ependymom fehlinterpretiert worden. Das immunhistochemische Expressionsprofil (kräftige häufig punktförmige Expression von Zytokeratin bei meist fehlender Expression des Epithelialen-Membran-Antigens (EMA, in Ependymomen positiv) und des Kaliumkanals Kir7.1 (in Plexuspapillomen und Plexuskarzinomen positiv) erlaubt jedoch in den meisten Fällen eine diagnostische Abgrenzung.[3]
Pathogenese
Über an der Pathogenese von papillären Tumoren der Pinealisregion beteiligte Mechanismen gibt es bisher keine Erkenntnisse.
Therapie
Der Tumor wird operativ entfernt. Insbesondere wenn eine vollständige Entfernung nicht möglich ist wird postoperativ eine Bestrahlung durchgeführt. Eine Behandlung sollte möglichst im Rahmen klinischer Studien erfolgen, die sich derzeit (2010) jedoch erst im Planungsstadium befinden.
Prognose
Auch nach vollständiger operativer Entfernung ist die Prognose von papillären Tumoren der Pinealisregion von häufigen Rezidiven geprägt.[2] Die biologische Wertigkeit von papillären Tumoren der Pinealisregion muss deshalb analog zu Grad II-III der WHO-Klassifikation der Tumoren des zentralen Nervensystems eingeschätzt werden.[2]
Literatur
Einzelnachweise
- ↑ Jouvet A: Papillary tumor of the pineal region. Am J Surg Pathol. 2003;27:505-12. PMID 12657936 (Erstbeschreibung)
- ↑ a b c Fevre-Montange M et al.: Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006;65:1004-11. PMID 17021405
- ↑ Hasselblatt M et al. Immunohistochemical profile and chromosomal imbalances in papillary tumours of the pineal region. Neuropathol Appl Neurobiol. 2006;32:278-83. PMID 16640646

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Tumor des zentralen Nervensystems
- Zirbeldrüse
Wikimedia Foundation.