- Gehirntumor
-
Ein Hirntumor ist eine Gewebswucherung des Gehirns. Tumoren der Hirnhäute, Meningeome, gehören zwar zu den intracraniellen Tumoren, also solchen innerhalb des knöchernen Schädels, zählen aber nicht zu den Hirntumoren im engeren Sinne. Da sie jedoch ab einer gewissen Größe immer auch Hirnstrukturen kompromittieren, werden sie (nicht ganz korrekt) bei Aufzählungen mit den Hirntumoren abgehandelt. Hirntumore können mehr oder weniger bösartig sein, setzen jedoch im allgemeinen keine Metastasen. Meistens entstehen sie aus dem Nervenstützgewebe (Gliome/Astrozytome) oder der Hypophyse oder es handelt sich um Tochtergeschwulste aus Tumoren anderer Organe (Hirnmetastasen).
Im Rahmen des selten auftretenden Turcot-Syndroms können Hirntumoren (hier meist Gliome) auch erblich bedingt sein und familiär gehäuft auftreten[1]. Ein weiteres vererbbares Syndrom, das mit Hirntumoren einhergehen kann, ist das Li-Fraumeni-Syndrom.
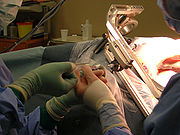 stereotaktische Probenentnahme
stereotaktische ProbenentnahmeInhaltsverzeichnis
Gutartige / bösartige Hirntumoren
Gutartige Tumoren verdrängen das umgebende Gewebe, wachsen jedoch nicht hinein und entwickeln keine Metastasen. Innerhalb des Schädels können sie jedoch z. B. durch Verlegung des Liquorsystems zu einem erhöhten Hirndruck führen. Da sie auch lebenswichtige Strukturen verdrängen können, ist der Begriff „gutartig“ hier irreführend und bezieht sich nur auf das histologische Kriterium, da diese Tumoren nicht in das umliegende Gewebe infiltrieren. Bei vollständiger Entfernung ist auch eine Heilung möglich. Abgesehen von einigen sehr speziellen Ausnahmen metastasieren Hirntumoren nie. Bösartige Hirntumoren wachsen infiltrierend, das heißt sie sind nicht scharf vom umliegenden Gewebe getrennt. Wenn nach einer Operation vereinzelte Zellen im Randbereich verbleiben und weiterwachsen, kann es zu einem Rezidiv kommen. Abhängig vom Malignitätsgrad kann dies schneller oder langsamer geschehen. Ein Beispiel für einen gutartigen Hirntumor ist das Akustikusneurinom. Ein Beispiel für einen bösartigen Tumor ist das Glioblastom.
Einteilung nach dem Ausgangsgewebe
Hirntumoren sind in je etwa einem Fünftel der Fälle Gliome, Meningeome und Hypophysenadenome. Ein weiteres Fünftel sind Metastasen von Tumoren aus anderen Körperteilen, die wie der Ausgangstumor in der jeweiligen Fachrichtung behandelt werden (49 % Bronchialkarzinom, 11 % Mammakarzinom). Relativ häufig ist auch das Akustikusneurinom.
Bei Erwachsenen finden sich häufig Tumoren, die ursprünglich aus anderen Organen stammen und im Gehirn Tochtergeschwülste (Metastasen) bilden. Der Anteil der Hirntumore an den bösartigen Tumoren im Erwachsenenalter beträgt 1 bis 4 %. Bei Kindern und Jugendlichen machen Tumoren, die ihren Ursprung direkt im Gehirn haben bis zu 20 bis 25 % aller bösartigen Neubildungen aus.
Neuroepitheliale Tumoren
- Astrozytäre Tumoren
- mit umschriebenem Wachstum
- pilozytisches Astrozytom, (WHO Grad I)
- pleomorphes Xanthoastrozytom, (WHO Grad II)
- subependymales Riesenzellastrozytom, (WHO Grad I)
- mit diffusem Wachstum
- diffuses Astrozytom (Varianten: fibrillär, protoplasmatisch, gemistozytisch), (WHO Grad II)
- anaplastisches Astrozytom (WHO Grad III)
- Glioblastom (Varianten: Gliosarkom, Riesenzell-Glioblastom), (WHO Grad IV)
- mit umschriebenem Wachstum
- Oligodendrogliale Tumoren
- Oligodendrogliom, (WHO Grad II)
- anaplastisches Oligodendrogliom, (WHO Grad III)
- Mischgliome
- Oligoastrozytom, (WHO Grad II)
- anaplastisches Oligoastrozytom, (WHO Grad III)
- Ependymale Tumoren
- myxopapilläres Ependymom
- Ependymom (Varianten: zellulär, papillär, tanzytisch, klarzellig)
- anaplastisches Ependymom
- Subependymom
- Plexus-Choroideus-Tumoren
- Gliome ungeklärter Abstammung
- Astroblastom
- Gliomatosis cerebri
- Chordoides Gliom des 3. Ventrikels
- Neuronale und gemischt neuronal–gliöse Tumoren
- Gangliozytom
- Dysplastisches Gangliozytom des Kleinhirns (Lhermitte-Duclos-Syndrom)
- Desmoplastisches infantiles Gangliogliom
- Dysembryoplastischer neuroepithelialer Tumor
- Gangliogliom
- Anaplastisches Gangliogliom
- Zentrales Neurozytom
- Zerebelläres Liponeurozytom
- Paragangliom des Filum terminale
- angiozentrisches Gliom
- glioneuronaler papillärer Tumor
- rosettenformender glioneuronaler Tumer des vierten Ventrikels
- Neuroblastäre Tumoren
- Olfaktoriusneuroblastom
- Olfaktoriusneuroepitheliom
- Pinealisparenchymtumoren
- Pineozytom
- Pineoblastom
- Pinealisparenchymtumor intermediärer Differenzierung
- Papillärer Tumor der Pinealisregion
- Embryonale Tumoren
- Medulloepitheliom
- Ependymoblastom
- Medulloblastom (Varianten: desmoplastisch, großzellig, melanotisch, Medullomyoblastom)
- Supratentorieller primitiver neuroektodermaler Tumor (PNET), Varianten: Neuroblastom, Ganglioneuroblastom)
- Atypischer teratoider/rhabdoider Tumor (AT/RT)
Meningeale Tumoren
- Meningeotheliale Tumoren
- Meningeom (Varianten: meningeothelial, fibroblastisch, transitionell, psammomatös, agniomatös, mikozystisch, sekretorisch)
- Atypisches Meningeom (Varianten: klarzellig, chordoid), (WHO Grad II)
- Anaplastisches Meningeom (Varianten: papillär, rhabdoid), (WHO Grad III)
- Mesenchymale, nicht-meningotheliale Tumoren
- Hämangiom
- Epitheloides Hämangioendotehliom
- Hämangioperizytom
- Angiosarkom
- Lipom
- Angiolipom
- Liposarkom
- malignes fibröses Histizytom
- Leiomyom
- Leiomyosarkom
- Rhabdomyosarkom
- Chondrom
- Chondrosarkom
- Osteom
- Osteosarkom
- Osteochondrom
- Kaposi-Sarkom
- Primäre melanozytäre Tumoren
- Melanozytose
- Melanozytom
- Malignes Melanom
- Meningeale Melanomatose
- Tumoren unsicherer Histogenese
- Hämangioblastom (Von-Hippel-Lindau-Erkrankung)
Lymphome und hämatopoetische Tumoren
- Malignes Lymphom
- Plasmozytom
- Granulozytäres Sarkom
Keimzelltumoren
- Germinom
- Embryonales Karzinom
- Dottersack-Tumor
- Chorionkarzinom
- Teratom (Varianten: reifes, unreifes, mit maligner Transformation)
- Gemischte Keimzelltumoren
Tumoren der Sellaregion
- Kraniopharyngeom (Varianten: adamantinös, papillär)
- Granularzelltumor
Metastasen
Supratentorielle – infratentorielle Hirntumoren
Diese Einteilung berücksichtigt, ob der Tumor oberhalb des Kleinhirnzeltes (tentorium cerebelli) oder unterhalb zu finden ist. Bei infratentoriellen Tumoren besteht die Gefahr, dass lebenswichtige Strukturen wie das Atemzentrum verdrängt oder infiltriert werden. Supratentorielle Raumforderung führen zu Massenverschiebung des Gehirns durch Tentoriumsschlitz in die hintere Schädelgrube. Folge: Kompression des Mittelhirns -> cranio-caudales Fortschreiten durch Verlust der Hirnstammfunktionen (= z.B Apallisches Syndrom)
Symptome
Die Symptome können vielseitig sein und lassen sich grob in vier Klassen einteilen: fokale neurologische Ausfälle (zum Beispiel Lähmungen, Gesichtsfeldausfälle) in Abhängigkeit von der Lokalisation, fokale Anfälle als Ausdruck einer symptomatischen Epilepsie, psychische Veränderungen (wie eine Verminderung des Antriebs) oder Folgen des Hirndrucks durch den Massenverdrängungseffekt (z. B. Kopfschmerzen, Übelkeit/Erbrechen oder Bewusstseinsstörung).
Diagnose
Sie wird heute in der Regel durch eine Magnetresonanztomographie (Kernspintomographie) gestellt. Hierbei kann eine Kontrastmittelaufnahme einen Hinweis auf die Malignität eines Tumors geben. Eine weitere Möglichkeit ist die Positronenemissionstomographie mit Methionin, einer Aminosäure mit radioaktivem Kohlenstoff, die eine erhöhte Mikrogefäßdichte anzeigt und somit ebenfalls einen Anhalt für Bösartigkeit liefert. Da diese Methode aufwändig ist und ein eigenes Zyklotron erfordert, ist sie nicht sehr verbreitet und nur großen Zentren vorbehalten.
Therapie
Die Therapie richtet sich nach der Lokalisation des Tumors, der Größe, dem Ursprungsgewebe und dem Allgemeinzustand des Patienten. Typischerweise steht bei höhergradigen Hirntumoren an erster Stelle eine Operation, gefolgt von einer Bestrahlung, teilweise in Kombination mit einer Chemotherapie, und einer nachfolgenden Chemotherapie. Bei niedriggradigen Tumor kann auch zunächst zugewartet werden.
Operation
Die Radikalität der Operation ist begrenzt durch das zu erwartende postoperative Defizit, das heißt man operiert so viel wie nötig und so wenig wie möglich. Bei vielen Hirntumoren ist das Tumorgewebe und das gesunde Hirngewebe während der OP nur schwer zu unterscheiden. Um dieses Risiko der Verletzung gesunden Gewebes zu verkleinern, werden verschiedene Methoden eingesetzt:
- Computergestützte Navigation
- Die Bilder der MRT und CT-Untersuchung werden in einem Computer zu einem räumlichen Modell verarbeitet. Während der OP werden Instrumente an einem speziellen Rahmen angebracht, der dem Computer die genaue Position der Instrumente übermittelt. Auf diese Weise kann der Neurochirurg während der OP an einem Monitor die Position seiner Instrumente kontrollieren.
- funktionelle Bildgebung
- Durch spezielle Untersuchungen (PET, fMRT) können Gehirnbereich und Funktion zugeordnet werden. Die um den Tumor liegenden Gehirnbereiche können schon vor der OP analysiert werden. Der Neurochirurg kann dann während der OP diese wichtigen Gehirnbereiche schonen.
- OP am wachen Patienten
- Während einer kurzen Narkose wird der Schädel geöffnet und das Gehirn freigelegt. Dann wird der Patient wieder erweckt. Da das Gehirn selbst über keine Schmerzsinneszellen verfügt, ist der Patient trotz der Wachheit schmerzfrei. Um vor Entfernung von Gehirngewebe sicher zu gehen, dass dieses Gehirngewebe keine wichtige Funktion hat, wird es vorher elektrisch stimuliert. Durch den harmlosen Stromstoß wird das Gehirngewebe gelähmt. Am wachen Patienten kann nun überprüft werden, ob es zu Lähmungen, Sehstörungen oder anderen neurologischen Ausfällen kommt.
- Fluoreszenzmarkierung
- Den Patienten wird vor der OP eine Substanz verabreicht, die nur von Tumorgewebe aufgenommen und abgebaut wird. Die Abbauprodukte fluoreszieren unter UV-Licht. Während der OP leuchtet das kranke Gewebe deutlich und kann entfernt werden.
Photonen-Strahltherapie
Die Bestrahlung kann zum einen von extern (also durch die Haut) fraktioniert erfolgen. Dabei werden zwei oder mehr Photonen-Strahlenquellen so ausgerichtet, dass sich die Strahlen im Zielgewebe kreuzen und das übrige Gewebe so wenig wie möglich schädigen. Fraktioniert bedeutet, dass man die Behandlung in bis zu 40 Sitzungen (je 5 Tage über 6 Wochen) aufteilt, damit sich das übrige Gewebe immer wieder erholen kann.
Eine andere Bestrahlungstechnik ist die Iod-Seed Implantation. Dabei wird eine Iod 125-Photonen-Strahlenquelle mit einer Stereotaktischen Hirnoperation in den Tumor eingebracht und bestrahlt sozusagen "von innen".
Ionen-Strahltherapie[2]
Die Bestrahlung erfolg mittels Ionen, z.B. Wasserstoff-, oder Kohlenstoff-Ionen, die in einem Teilchenbeschleuniger auf hohe Geschwindigkeiten beschleunigt wurden, siehe Partikeltherapie bzw. Schwerionentherapie. Diese Methode hat gegenüber der Photonen-Strahltherapie zwei entscheidende Vorteile :
- Die Absorption der Ionen außerhalb des Zielgebietes ist vergleichsweise gering, der größte Teil der Strahlenergie wird im Tumor abgegeben. Die Schädigung des umgebenden Gewebes bleibt also gering.
- Sowohl die Eindringtiefe als auch die Position in der Bestrahlungsebene können schnell und präzise variiert werden, wodurch auch komplexe Tumorgeometrieen erfasst werden können.
Chemotherapie
Für die Chemotherapie gibt es verschiedene Schemata. Die heutzutage am häufigsten angewendeten sind ACNU+VM26, Fotemustin, Temozolomid und PCV.
PCV steht für Procarbazin, Cecenu und Vincristin, ein Zyklus dauert 6 Wochen. Die Medikamente werden teilweise als Tabletten und teilweise als Infusionen über fast den gesamten Zeitraum eingenommen. Die Nebenwirkungen sind häufig und ausgeprägt. Die Dosis einer Chemotherapie ist abhängig von der Körperoberfläche, die mit der Formel von DuBois und DuBois aus Größe und Gewicht berechnet wird.
![\mathrm{KOF} = \frac {\mathrm{Gewicht} ^{0,425} {\,}[\mathrm{kg}] \cdot \mathrm{Groesse} ^{0,725} {\,}[\mathrm{cm}] \cdot 71,84 {\,}[\mathrm{m}^2 /\mathrm{kg} \cdot \mathrm{cm}] } { 1000}](/pictures/dewiki/97/a33cf71001cb9266d5174797e6106242.png)
Prognose
Je bösartiger ein Hirntumor, umso schlechter ist die Prognose. Tumoren nach WHO-Grad I sind prinzipiell heilbar, Grad-II-Tumoren haben eine relativ gute Prognose, wenn sie nicht malignisieren, das heißt im Verlauf bösartig werden.
Tumoren nach WHO-Grad III und insbesondere Grad IV (Glioblastom) haben unbehandelt eine sehr schlechte Prognose von nur wenigen Monaten. Obwohl sich die Prognose durch die etablierten Therapien deutlich verbessert hat und sich jetzt im Bereich von 1-2 Jahren bewegen kann, ist sie weiterhin schlecht. Das Thema Prognose ist gerade bei bösartigen Erkrankungen als sehr heikel anzusehen, da Überlebenszeiten immer Mittelwerte von Patienten-Kollektiven darstellen und immer auch abhängig sind vom frühen oder späten Zeitpunkt einer Diagnosestellung.
Im Einzelfall kann man einem Patienten keine Vorhersage geben, ob er länger oder kürzer als die in Studien ermittelte mittlere Überlebenszeit leben wird.
Tabelle: WHO-Gradierung von Tumoren des Nervenzentrums und Prognose
WHO-Grad Tumor-Dignität Histologische Charakteristika Prognose (Mittlere Überlebenszeit Zirka-Angaben) Grad I Benigne Gut differenziertes Gewebe, keine Metastasen >5(-50) Jahre Grad II Semibenigne Einzelne atypische Zellen, noch gut differenziertes Gewebe, Kernatypien, keine/kaum Metastasen 3-5 Jahre Grad III Semimaligne Viele atypische Zellen, Mitosen, Ursprungsgewebe noch erkennbar, jedoch bereits entdifferenziert 2-3 Jahre Grad IV Maligne Entdifferenziertes Gewebe, viele Mitosen, Nekrosen, Endothelproliferation, Metastasen 6 Monate (bis 2 Jahre) Folgende histologische Kriterien werden zur Beurteilung herangezogen: a)Kernatypien, b)Mitosen, c)Endothelproliferation, d)Nekrosen. Wenn keines der Kriterien erfüllt ist, Grad I. 1 Kriterium erfüllt, Grad II. 2 Kriterien vorhanden, Grad III. 3 oder 4 Kriterien vorhanden, Grad IV.
Siehe auch
Einzelnachweise
- ↑ gms: Die Bedeutung des Glioblastoms im Rahmen des HNPCC-Screenings. 7. Oktober 2004.
- ↑ HIT: Heidelberger Ionenstrahl-Therapie 19.05.2008
Weblinks
- uni-duesseldorf.de – Leitlinien zu Hirntumoren im Kindesalter

Bitte beachte den Hinweis zu Gesundheitsthemen! - Astrozytäre Tumoren
Wikimedia Foundation.