- Real time pcr
-
Die Real-Time-quantitative-PCR (kurz RTq-PCR oder qRT-PCR, auch Real Time Detection PCR, kurz RTD-PCR) ist eine Vervielfältigungsmethode für Nukleinsäuren, die auf dem Prinzip der herkömmlichen Polymerase-Kettenreaktion (PCR) beruht, und zusätzlich die Quantifizierung der gewonnenen DNA ermöglicht. Die Quantifizierung wird mit Hilfe von Fluoreszenz-Messungen durchgeführt, die während eines PCR-Zyklus erfasst werden (daher der Name „Real Time“). Die Fluoreszenz nimmt proportional mit der Menge der PCR-Produkte zu. Am Ende eines Laufs (der aus mehreren Zyklen besteht) wird anhand von erhaltenen Fluoreszenzsignalen die Quantifizierung in der exponentiellen Phase der PCR vorgenommen. Nur in der exponentiellen Phase der PCR (die wenige Zyklen in einem Lauf dauert) ist die korrekte Quantifizierung möglich, da während dieser Phase die optimalen Reaktionsbedingungen herrschen. Diese Methode unterscheidet sich somit von anderen quantitativen PCR-Methoden (qPCR), die erst nach Ablauf der PCR eine quantitative Auswertung (z. B. Kompetitive PCR), meist unter Einbeziehung einer gelelektrophoretischen Auftrennung der PCR-Fragmente, vornehmen.
Die Real-Time-quantitative-PCR kann auch für andere Zwecke verwendet werden, z. B. zur Unterscheidung zwischen homozygoten und heterozygoten Zellen.
Für die Real-Time-PCR wird manchmal die Abkürzung RT-PCR verwendet, dies führt aber zu Verwechslungen, da diese Abkürzung bereits für die Reverse-Transkriptase-Polymerasekettenreaktion verwendet wird. Auch die Abkürzung qRT-PCR kann zu Verwechslungen führen, da auch die quantitative Reverse-Transkriptase-Polymerasekettenreaktion so abgekürzt wird.
Inhaltsverzeichnis
Methoden
Interkalierende Farbstoffe
Die einfachste Möglichkeit der Quantifizierung der PCR-Produkte ist die Nutzung von DNA-Farbstoffen (z. B. Ethidiumbromid oder SYBR Green I).
Diese Fluoreszenzfarbstoffe lagern sich in die DNA ein (interkalieren) bzw. binden an die doppelsträngige DNA, wodurch die Fluoreszenz dieser Farbstoffe ansteigt. Die Zunahme der Target-DNA korreliert daher mit der Zunahme der Fluoreszenz von Zyklus zu Zyklus. Die Messung findet am Ende der Elongation in jedem Zyklus statt.
Ein Nachteil dieses Verfahrens ist die geringe Spezifität, da zwischen verschiedenen PCR-Produkten nicht unterschieden werden kann. Außerdem können keine Multiplex-Messungen durchgeführt werden.
Den ersten Nachteil kann man ausgleichen, indem man nach abgelaufener PCR eine Schmelzkurvenanalyse durchführt, anhand derer die Fragmentlänge(n) und dadurch die Spezifität bestimmt werden kann.
Bei einer Schmelzkurvenanalyse wird die DNA aufgeschmolzen, indem die Temperatur langsam kontinuierlich erhöht wird (50 °C → 95 °C). Bei einer für das Fragment spezifischen Schmelztemperatur denaturiert der Doppelstrang zu zwei einzelsträngigen Molekülen. Dabei wird der Fluoreszenzfarbstoff (z. B. SYBR Green I) freigesetzt, und es wird eine Fluoreszenzabnahme registriert. Da die doppelsträngige DNA von spezifischen PCR-Produkten einen höheren Schmelzpunkt hat als unspezifisch entstehende Primerdimere, ist eine Unterscheidung möglich. Die Höhe des Peaks der Schmelzkurve gibt annähernd Auskunft über die Menge des gebildeten Fragments.
FRET-Sonden
Eine andere Möglichkeit ist, den Fluorescence resonance energy transfer (FRET) auszunutzen. Ein Donor-Fluorochrom (Reporter - im Zusammenhang mit TaqMan Sonden), das durch eine Lichtquelle angeregt wird, gibt einen Teil seiner Energie an ein in ausreichender Nähe befindliches Akzeptor-Fluorochrom (bzw. einen dark Quencher - im Zusammenhang mit TaqMan Sonden) ab. Nimmt der Abstand zwischen Akzeptor und Donor zu, so nimmt FRET und somit das Fluoreszenzsignal des Akzeptors ab, während das des Donors zunimmt. Diese Methode ist sehr aufwendig und teuer, bietet aber die Vorteile der hohen Spezifität des Assays.
LightCycler-Sonden (auch Hybridisierungs-Sonden)
Die einfachste Möglichkeit der Nutzung des FRET zur Quantifizierung von Nukleinsäuren besteht in der Verwendung von LightCycler-Sonden. Zwei verschiedene, jeweils mit einem FRET-Donor bzw. FRET-Akzeptor (hier Reporter) markierte Oligonukleotide, die nebeneinander an die Ziel-Sequenz binden und damit die Fluorochrome in eine für den FRET ausreichende Nähe bringen, können als Sonden für die Quantifizierung der PCR-Produkte eingesetzt werden. Die Messung findet am Ende der Annealing-Phase in jedem Zyklus statt. Auch hier kann sich eine Schmelzkurvenanalyse anschließen.
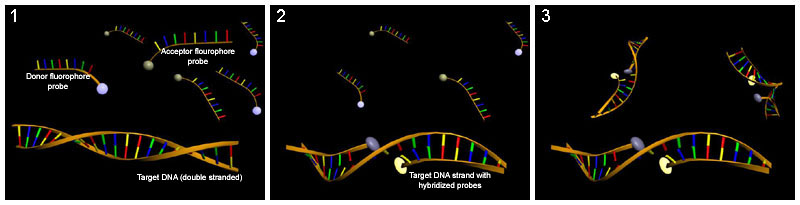 Quantifizierung von Nucleinsäuren mit Hilfe der Real-time-PCR und LightCycler-Sonden: (1) Einsatz von LightCycler-Sonden, die mit 2 verschiedenen FRET-Fluorophoren markiert wurden. (2) Während eines PCR-Zyklus hybridisieren die Sonden mit dem komplementären DNA-Strang und ermöglichen somit eine Fluoreszenz des Akzeptors. (3) Die Akzeptor-Fluoreszenz (hier der Reporter) steigt proportional mit der Konzentration komplementärer DNA.
Quantifizierung von Nucleinsäuren mit Hilfe der Real-time-PCR und LightCycler-Sonden: (1) Einsatz von LightCycler-Sonden, die mit 2 verschiedenen FRET-Fluorophoren markiert wurden. (2) Während eines PCR-Zyklus hybridisieren die Sonden mit dem komplementären DNA-Strang und ermöglichen somit eine Fluoreszenz des Akzeptors. (3) Die Akzeptor-Fluoreszenz (hier der Reporter) steigt proportional mit der Konzentration komplementärer DNA.TaqMan-Sonden (auch Hydrolyse-Sonden)
Eine weitere häufig genutzte Möglichkeit des FRET besteht in der Anwendung einer Sonde, die an ihrem einen Ende mit dem Quencher, an ihrem anderen Ende mit einem Reporter-Fluoreszenzfarbstoff (z. B. TAMRA und FAM) markiert wird (Double-Dye-Oligos, TaqMan-Sonde). Wenn die Taq-Polymerase, die zusätzlich zur Polymeraseaktivität eine 5'-3'-Exonuclease-Aktivität besitzt, die Sonde während der Synthese des Gegenstranges am 5'-Ende abbaut, entfernen sich dadurch Quencher und Fluorophor voneinander, und eine steigende Reporter-Fluoreszenz kann gemessen werden. Die Messung findet am Ende der Elongation in jedem Zyklus statt.
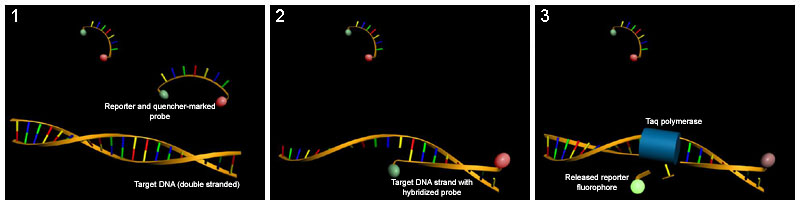 Quantifizierung von Nucleinsäuren mit Hilfe der Real-time-PCR und TaqMan-Sonden. (1) Die Fluoreszenz des Reporter-Fluorophors wird bei intakten TaqMan-Sonden durch einen Quencher durch strahlungsfreie Energieübertragung (FRET) unterdrückt. (2) Während eines PCR-Zyklus hybridisiert die Sonde mit dem komplementären DNA-Strang, die Reporter-Fluoreszenz bleibt zunächst unterdrückt. (3) Die Taq-Polymerase baut auf Grund ihrer 5'-3'-Exonucleaseaktivität das 5'-Ende der Sonde während der PCR-Zyklen ab. Die Fluoreszenz des Reporters wird nun nicht mehr durch den Quencher gelöscht und kann gemessen werden.
Quantifizierung von Nucleinsäuren mit Hilfe der Real-time-PCR und TaqMan-Sonden. (1) Die Fluoreszenz des Reporter-Fluorophors wird bei intakten TaqMan-Sonden durch einen Quencher durch strahlungsfreie Energieübertragung (FRET) unterdrückt. (2) Während eines PCR-Zyklus hybridisiert die Sonde mit dem komplementären DNA-Strang, die Reporter-Fluoreszenz bleibt zunächst unterdrückt. (3) Die Taq-Polymerase baut auf Grund ihrer 5'-3'-Exonucleaseaktivität das 5'-Ende der Sonde während der PCR-Zyklen ab. Die Fluoreszenz des Reporters wird nun nicht mehr durch den Quencher gelöscht und kann gemessen werden.Molecular Beacons
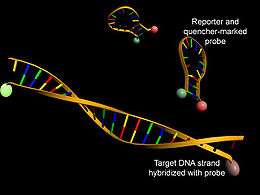 Real time quantitative PCR mit Hilfe von Molecular Beacons als Sonden.
Real time quantitative PCR mit Hilfe von Molecular Beacons als Sonden.Eine weitere Möglichkeit der Echtzeit-Quantifizierung von PCR-Produkten unter Ausnutzung des FRET bietet die Nutzung von Molecular Beacons als Sonden. Molecular Beacons sind Oligonucleotide, die sowohl mit einem Reporter-Fluorophor als auch mit einem Quencher gekoppelt sind. Die Nucleotide am 5'-Ende der Sonde sind zu denen am 3'-Ende komplementär, so dass sich eine für Molecular Beacons charakteristische Sekundärstruktur ausbilden kann. In diesem als stem loop (Haarnadelstruktur) bezeichneten Zustand zeigt der Reporter durch seinen geringen Abstand zum Quencher keine Fluoreszenz. Durch Anlagerung der Schleifen-Region an eine komplementäre DNA-Sequenz während eines PCR-Zyklus wird der Abstand zwischen Quencher und Reporter vergrößert. Eine Reporter-Fluoreszenz kann somit beobachtet werden.
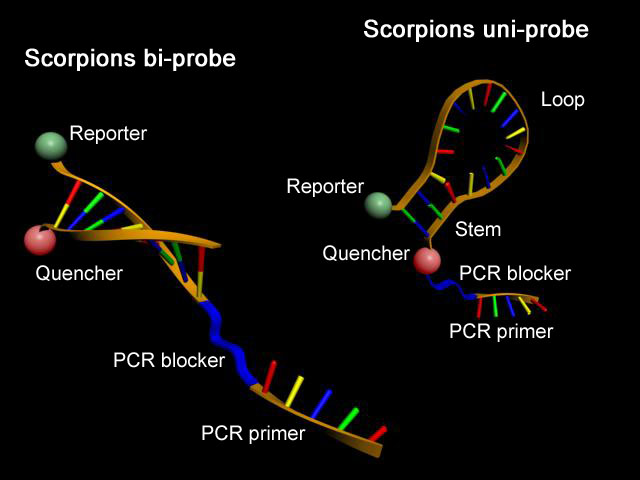
Scorpion-Primer
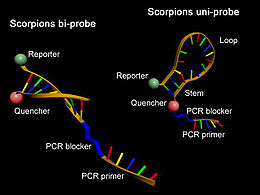 Scorpion-Primer.
Scorpion-Primer.Scorpion-Primer sind komplexe Oligonucleotide, welche die Eigenschaften von Real-Time-PCR-Sonden und PCR-Primern in einem (Uni-Scorpion) oder zwei Molekülen (Bi-Scorpion) vereinigen. Ähnlich den Molecular Beacons besitzen sie eine charakteristische Sekundärstruktur mit einer selbstkomplementären Schaft-Region, deren Enden mit einem Reporter-Fluorophor und einem Quencher modifiziert wurden. Zusätzlich tragen diese Sonden am 3'-Ende einen PCR-Primer. Während eines PCR-Zyklus kann mit steigender DNA-Konzentration eine Reporter-Fluoreszenz durch Anlagerung der Loop-Region an eine komplementäre DNA-Sequenz und damit vergrößerten Abstand zwischen Quencher und Reporter beobachtet werden.
Lux-Primer
Lux-Primer sind mit einem Fluoreszenzfarbstoff markierte Oligonucleotide, deren Fluoreszenzintensität von ihrer jeweiligen chemischen Umgebung abhängt. Werden diese Primer während einer PCR in eine DNA eingebaut, kann eine Zunahme der Fluoreszenz beobachtet werden.
Quantifizierung
Verschiedene Rechenmodelle werden für die Quantifizierung herangezogen, wobei meistens ein Referenz-Gen (z. B. GAPDH, Actin, Tubulin) mitgemessen wird, um einen relativen Menge-Vergleich durchzuführen (relative Quantifizierung). Andere, weitaus kompliziertere Methoden sollen eine absolute Quantifizierung ermöglichen, bei der die genaue Anzahl der in der Probe vorhandenen Templates bestimmt werden kann.
Ct-Wert oder auch Cp-Wert
In der ersten Phase der Amplifikation einer PCR ist die Templatemenge begrenzt und die Wahrscheinlichkeit, dass sich Template, Primer und Polymerase treffen, suboptimal, während in der dritten Phase der Amplifikation die Menge der Produkte (DNA, Pyrophosphat, Monophosphatnucleotide) derart ansteigt, dass es zur Hemmung durch diese kommt, häufiger Produktfragmente miteinander hybridisieren, die Substrate langsam verbraucht werden und letztlich die Polymerasen und Nucleotide durch die Hitze langsam zerstört werden. Ein exponentieller und daher quantifizierbarer Anstieg findet sich nur in der Phase dazwischen. Exponentiell bleibt eine PCR bei 12 bis 400 Ausgangskopien für ca. 30 Zyklen, bei 200 bis 3200 für 25 Zyklen und bei anfänglich 3200 bis 51200 für höchstens 20 Zyklen. Um immer am Anfang der exponentiellen Phase messen zu können, wird häufig der Ct-Wert (engl. Cycle Threshold für Schwellenwert-Zyklus) bzw. der Cp-Wert (engl. Crossing Point) verwendet, der den Zyklus beschreibt, an dem die Fluoreszenz erstmalig signifikant über die Hintergrund-Fluoreszenz ansteigt.
Effizienz
Die Effizienz kann auf verschiedene Arten berechnet werden, die sich in ihrem Ergebnis leicht unterscheiden. Die einfachste Art ist folgende:
Die Effizienz E kann mit Hilfe der Steigung m einer Standardkurve berechnet werden:[1]
- E = 10 − 1 / m
Eine Steigung m von −3,32 würde somit eine Effizienz von 100 % bedeuten, d. h. eine Verdopplung der Amplifikate pro Zyklus. -3.58 eine Effizienz von 90 %.
Absolute Quantifizierung
Eine absolute Quantifizierung ist aufwändig und die Ergebnisse fragwürdig, daher wird diese Art der Quantifizierung selten durchgeführt. So muss u. a. die Effizienz der reversen Transkription, die zwischen 5 und 95 % liegen kann, bestimmt werden, z. B. durch Verwendung synthetisierter RNA bekannter Menge.
Relative Quantifizierung
Hierfür wird eine interne Kontrolle benötigt. Eine interne Kontrolle kann ein Gen-Transkript sein, dessen Signal verwendet wird, um Variationen in der Ausgangsmenge der eingesetzten RNA auszugleichen. Dies wird als Normierung bezeichnet. Weil die Gesamtanalyse auf diesem Signal beruht, ist die Wahl der internen Kontrolle ein wichtiger Aspekt des Experiments. Die ideale interne Kontrolle ist leicht zu detektieren,und deren Expression sollte nicht während des Zellzyklus, zwischen Zelltypen oder als Antwort auf die experimentelle Behandlung (z. B. Stress, Medikamente, Krankheit) variieren.
Berechnung mit Hilfe einer Standard-Kurve
Es besteht eine lineare, umgekehrt proportionale Beziehung zwischen dem Logarithmus der eingesetzten Menge und dem Ct. Ist die Ausgangsmenge bekannt, kann eine Standardkurve durch Auftragen des Logarithmus der Ausgangsmenge gegen den Ct konstruiert werden. Durch die Geradengleichung
- x = (Ct − b) / m
kann an der Standardkurve für jede unbekannte Probe der Logarithmus der Kopienzahl bestimmt werden. Alle Proben werden normiert, indem die errechnete Kopienzahl des Targetgens durch die Kopienzahl der internen Referenz geteilt wird:
- Gen (normiert) = Kopienzahl Target / Kopienzahl Referenz
Die unterschiedliche Expression zweier Proben relativ zueinander lässt sich als Quotient darstellen und ergibt eine n-fache Expression:
- Gen (normalisiert) (Gruppe A) / Gen (normalisiert) (Gruppe B) = n-fache Expression Gruppe A zu Gruppe B
Berechnung nach der ΔΔCt-Methode
Die unterschiedliche Expression wird als n-fache Expression mit Hilfe des ΔΔCt-Wertes angegeben. Wichtig bei diesem Verfahren ist eine gleiche Effizienz der beiden beteiligten PCR-Reaktionen. Die Ct-Werte werden hierbei einfach voneinander abgezogen (ΔCt), die beiden ΔCt-Werte der einzelnen Gruppen (z. B. krank/gesund, mit/ohne Wirkstoff) voneinander abgezogen (ΔΔCt-Wert) und in die Gleichung n-fache Expression (Gruppe A zu Gruppe B) = 2 −ΔΔCt eingesetzt.
Neue Quantifizierungsalgorithmen
Um die Genauigkeit der relativen Quantifizierung zu verbessern, sind verschiedene, teils auf der ΔΔCt-Methode basierende Ansätze entwickelt worden.
- Erweiterung der Formel der ΔΔCt-Methode um die Effizienz des jeweiligen PCR-Ansatzes, die zuvor in einem Probelauf über eine Standardkurve bestimmt werden muss.
- Mittels Regressionsanalysen Fit der Real-time-PCR-Datensätze an eine exponentielle Funktion bzw. in linearisierter Form an eine Geradengleichung. Der Schnittpunkt dieser Funktionen mit der y-Achse gibt dann Aufschluss über die ursprünglich im Ansatz vorhandene DNA-Menge (relative Angabe im Vergleich zu einer Referenzprobe).
- Fit der Real-time-PCR-Daten an eine drei- oder mehrparametrige sigmoide Funktion. Auch hier wird die relative DNA-Menge über y(0) bestimmt.
Vorteile dieser neuen, überwiegend noch nicht marktreifen Methoden liegen in der genaueren Analyse und geringeren Varianz der PCR-Ergebnisse. Bei einigen Methoden wird die Effizienz in jedem einzelnen Probenansatz berücksichtigt und ermöglicht so eine single tube Analyse.
Reproduzier- und Vergleichbarkeit der Ergebnisse
Ein Experiment ist gar nichts wert, wenn man es nicht wiederholen kann. Hierfür ist es absolut notwendig die Versuchsbedingungen konstant zu halten. Dabei ist hier nicht nur die Konsistenz der Assay-Qualität (Primer, Sonden, Polymerase, Puffer etc.) zu berücksichtigen, auch die Geräte müssen den Anforderungen genügen. Beispielsweise ist bei Block-Systemen (96- oder 384-Wells) die so genannte Homogenität das große Kriterium. Die Assays müssen in jedem Well des Blocks und zudem auf Blöcken baugleicher Geräte gleichgut laufen. Weiterhin darf sich im zeitlichen Verlauf diese Homogenität nicht verändern. Um sicher zu stellen, ob man mit dem Gerät im Labor sichere Experimente durchführen kann, sollte man in regelmäßigen Abständen entsprechende Kontrollen durchführen (einige Modelle neigen extrem zum Altern!). Dabei wird ein Assay in vielen Replikaten über den gesamten Block verteilt analysiert. Ganz klar: das Ergebnis sollte im Idealfall überall gleich sein. Jetzt sollte dem Experimentator bewusst sein, inwieweit Abweichungen auch relevante Auswirkungen haben können. Diese Abweichungen lassen sich übrigens nicht mittels teurer Wiederholungen ausgleichen, da dieser ebenso konstant wiederholt werden würde.
Einzelnachweise
Literatur
- Bianca Holzapfel & Lucia Wickert (2007): Die quantitative Real-Time-PCR (qRT-PCR). In: Biologie in unserer Zeit. Band 37, Nr. 2, S. 120-126. doi:10.1002/biuz.200610332
- Michael Walter Pfaffl (2004): Real-time RT-PCR: Neue Ansätze zur exakten mRNA Quantifizierung. In: Biospektrum. PDF
Weblinks
- www.gene-quantification.info englische Seite mit allem Wissen, Tips und Tricks über qPCR
- Real Time PCR Tutorial
- realtimepcr.dk Real time PCR (Kopenhagen)
Wikimedia Foundation.