- STED-Mikroskop
-
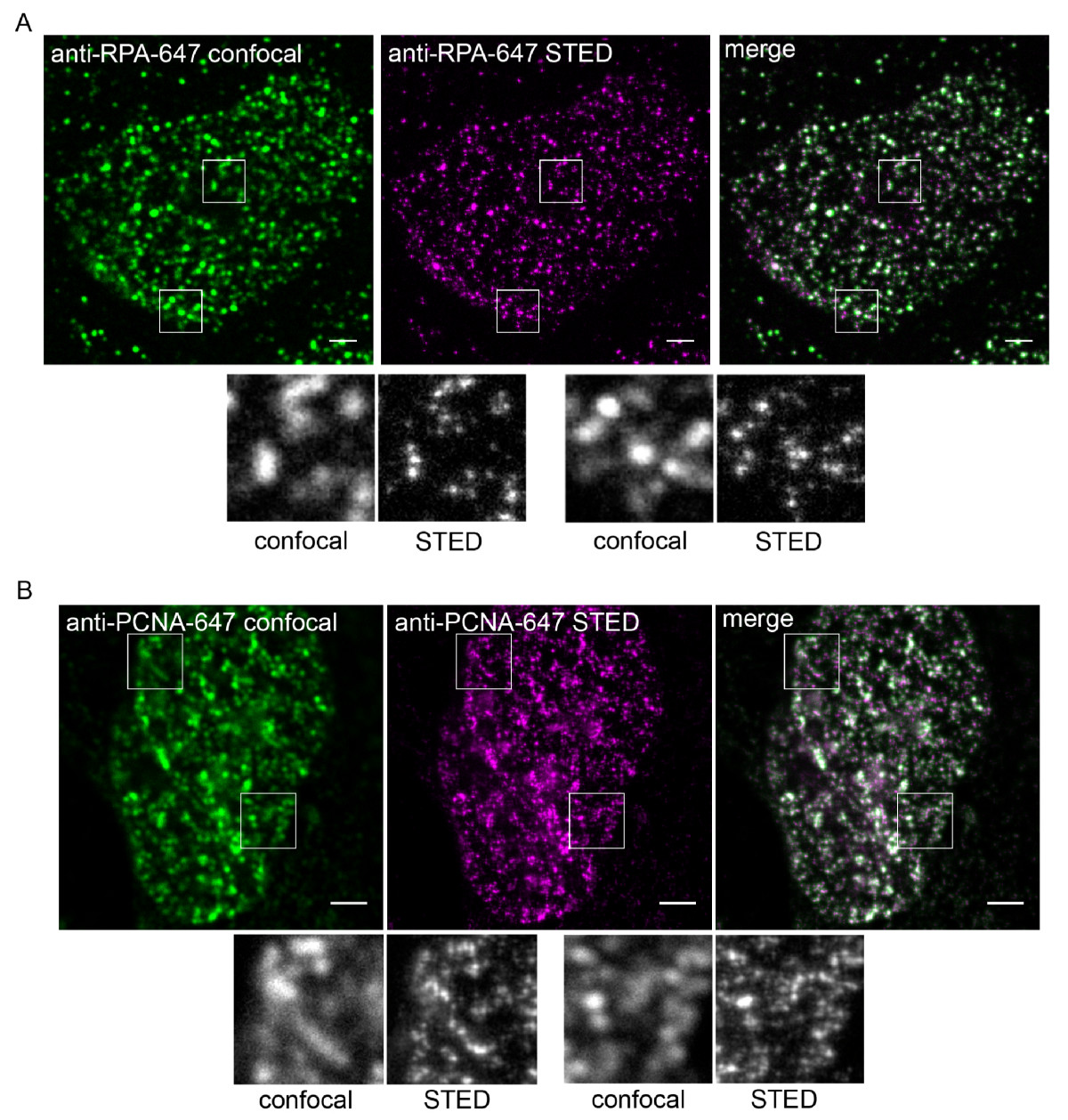
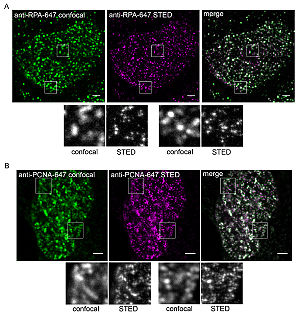 Vergleich von Standard-Konfokalmikroskopie (grüne Bilder, links) und STED-Mikroskopie (violette Bilder, Mitte) bei der Abbildung von DNA Replication factories, Protein-Komplexen zur DNA-Replikation, im Zellkern menschlicher Zellen. Die durch Überlagerung der Konfokal- und der STED-Aufnahmen erstellten Bilder (rechts) sowie die Ausschnitte (kleine graue Bilder) verdeutlichen die höhere Auflösung der STED-Mikroskopie.
Vergleich von Standard-Konfokalmikroskopie (grüne Bilder, links) und STED-Mikroskopie (violette Bilder, Mitte) bei der Abbildung von DNA Replication factories, Protein-Komplexen zur DNA-Replikation, im Zellkern menschlicher Zellen. Die durch Überlagerung der Konfokal- und der STED-Aufnahmen erstellten Bilder (rechts) sowie die Ausschnitte (kleine graue Bilder) verdeutlichen die höhere Auflösung der STED-Mikroskopie.
Ein STED-Mikroskop (STED = Stimulated Emission Depletion) ist ein Fluoreszenzmikroskop, dessen Auflösung nicht beugungsbegrenzt ist und das nach dem RESOLFT-Prinzip arbeitet. Es wurde 1994 von Stefan Hell theoretisch beschrieben[1] und 1999 von ihm experimentell realisiert[2]. Seitdem wird es unter anderem in seiner Arbeitsgruppe am Max-Planck-Institut für biophysikalische Chemie in Göttingen weiterentwickelt.
Aufgrund von Beugung ist die Auflösung herkömmlicher Fernfeldmikroskope begrenzt. Es lassen sich daher keine Details auflösen, die kleiner als circa die halbe Wellenlänge des verwendeten Lichts sind. Konfokalmikroskope können daher nebeneinanderliegende Strukturen nur bis zu einer Größe von ca. 200 nm auflösen. Die Auflösung für hintereinanderliegende Objekte (Tiefenauflösung) ist deutlich schlechter. Beim STED-Mikroskop wird durch gezieltes Ausschalten von Fluoreszenzfarbstoffen die Auflösung über die Beugungsgrenze hinaus gesteigert. Es konnte bereits eine Auflösung von 5,8 nm (lateral) gezeigt werden.[3]
Das STED-Mikroskop und die Gruppe um Stefan Hell wurden für ihre Ergebnisse im Jahr 2006 mit dem Deutschen Zukunftspreis ausgezeichnet.
Inhaltsverzeichnis
Grundlagen
Ein STED-Mikroskop baut auf Fluoreszenz-Laser-Raster-Mikroskopen auf. Um das Funktionsprinzip besser zu verstehen, ist es notwendig, auf die wichtigsten Gesichtspunkte von Fluoreszenz, stimulierter Emission und der Laser-Raster-Mikroskopie einzugehen.
Fluoreszenz und Stimulierte Emission
Bei der STED-Mikroskopie werden sogenannte Fluoreszenzfarbstoffe zum Markieren einzelner Bereiche eines Präparats eingesetzt. Solche Farbstoffe können durch Licht bestimmter Wellenlängen (Farben) „angeregt“ werden: Sie absorbieren ein Photon und gehen in einen energiereicheren Zustand über. Aus diesem Zustand können sie nach kurzer Zeit spontan durch Aussenden eines Photons größerer Wellenlänge (anderer Farbe) wieder in den Grundzustand zurückkehren. Diese spontane Abstrahlung von Licht nennt man Fluoreszenz. Durch geeignete Farbfilter kann das Fluoreszenzlicht vom Anregungslicht getrennt werden.
Ein angeregtes Farbstoffmolekül kann außer durch Fluoreszenz auch durch stimulierte Emission wieder in den Grundzustand zurückkehren. Dies passiert, wenn das angeregte Farbstoffmolekül mit Licht von ungefähr der gleichen Wellenlänge wie der des Fluoreszenzlichts bestrahlt wird. Das angeregte Farbstoffmolekül kann so zum sofortigen Übergang in den Grundzustand durch Aussenden eines Photons exakt derselben Wellenlänge stimuliert werden. Spontanes Fluoreszenzlicht kann dann nicht mehr ausgesandt werden, das Molekül ist ja nicht mehr im angeregten Zustand. Fluoreszenzmoleküle können also durch Stimulierte Emission ausgeschaltet werden: Wenn sie zur Emission des Lichtes stimuliert werden, können sie danach kein spontanes Fluoreszenzlicht mehr abgeben. Spontanes Fluoreszenzlicht und Licht von der Stimulierten Emission können z. B. durch Farbfilter voneinander getrennt werden.
Fluoreszenz-Laser-Raster-Mikroskopie
Für die Untersuchung in einem Fluoreszenzmikroskop werden Fluoreszenzfarbstoffe an bestimmte Stellen des zu untersuchenden Präparats gebracht. Wird das Präparat nun mit Licht geeigneter Wellenlänge beleuchtet, so werden die Farbstoffe zur Fluoreszenz angeregt und man erhält ein Bild der Farbstoffverteilung im Präparat. Im Laser-Raster-Mikroskop wird nicht das ganze Präparat auf einmal beleuchtet, sondern ein Laserstrahl („Anregungsstrahl“) wird lediglich auf einen kleinen Punkt in dem Präparat fokussiert. Die in diesem Bereich angeregte Fluoreszenz wird detektiert. Durch Bewegen (Scanning, Rastern) des Fokus über das Präparat wird punktweise ein Abbild der gefärbten Bereiche erstellt. Die Größe des Fokus bestimmt die maximale Feinheit der Details, die in dem Präparat gerade noch aufgelöst (getrennt wahrgenommen) werden können. Aufgrund der Beugung kann der Fokus nicht beliebig klein gewählt werden. Ein Laserstrahl lässt sich nicht auf einen Fleck kleiner als circa seine halbe Wellenlänge fokussieren.
Funktionsprinzip des STED-Mikroskops
 Anregungsfokus (links), ringförmiger Ausschaltefokus (mitte) und verbleibender fluoreszierender Bereich (rechts).
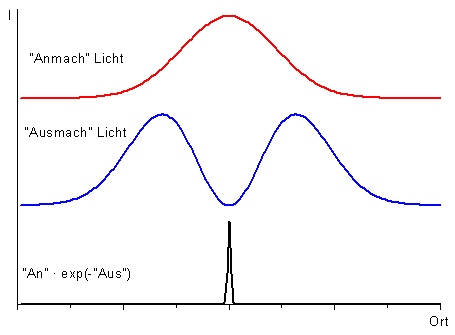
Anregungsfokus (links), ringförmiger Ausschaltefokus (mitte) und verbleibender fluoreszierender Bereich (rechts).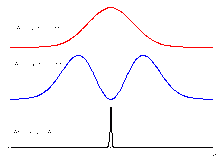 Querschnitt durch die Intensitätsprofile: Oben: Anregung, Mitte: Ausschaltelicht, Unten: Verbleibende Fluoreszenz
Querschnitt durch die Intensitätsprofile: Oben: Anregung, Mitte: Ausschaltelicht, Unten: Verbleibende FluoreszenzMit einem STED-Mikroskop ist eine bessere Auflösung als in einem herkömmlichen Laser-Raster-Mikroskop möglich: Der Bereich, aus dem Fluoreszenz emittiert wird, wird dabei bedeutend kleiner gemacht als der Bereich, der von dem Laserstrahl beleuchtet wird. Das wird durch gezieltes Ausschalten der Farbstoffmoleküle im Außenbereich des Fokus erreicht. Dazu wird das Präparat nicht nur mit dem fokussierten Anregungsstrahl beleuchtet (linkes Bild), sondern gleichzeitig mit einem zweiten Laserstrahl, dem „Ausschaltestrahl“. Diesem Ausschaltestrahl gibt man ein ringförmiges Profil im Fokus (mittleres Bild). In der Mitte, also dort wo der Anregungsstrahl seine maximale Helligkeit hat, ist der Ausschaltestrahl vollkommen dunkel. Der Ausschaltestrahl beeinflusst also die Fluoreszenzfarbstoffe in der Mitte nicht. Er schaltet aber die Fluoreszenzfarbstoffe im Außenbereichs des Anregungsfokus durch stimulierte Emission (siehe oben) aus; die Farbstoffmoleküle im Außenbereich bleiben dunkel, obwohl sie von dem Anregungslaser beleuchtet werden. Es leuchten deshalb nur die Farbstoffmoleküle genau aus dem Zentrum (rechtes Bild). Wenn der Ausschaltestrahl eine hohe Intensität hat (sehr hell ist), ist dieser Bereich sehr viel kleiner als der mit dem Anregungslaser beleuchtete Bereich (siehe Linienprofile rechts). Beim Scannen des Präparates erfasst man somit jeweils einen leuchtenden Fleck, der viel kleiner ist als in einem normalen Laser-Raster-Mikroskop. Deshalb kann man feinere Details auflösen. Um ein vollständiges Bild zu erhalten, wird das Präparat Punkt für Punkt abgerastert.
Die Größe des resultierenden Lichtflecks sinkt mit steigender Intensität des Ausschaltestrahls immer mehr ab. Das bedeutet, die Auflösung steigt immer mehr an je heller der Ausschaltestrahl ist und es gibt prinzipiell keine Grenze mehr für die erreichbare Auflösung.[4] Vor der Erfindung der STED-Mikroskopie bestand das Problem, dass der Anregungsstrahl aufgrund der Abbeschen Beugungsgrenze nicht beliebig klein fokussiert werden kann. Man regt also immer alle Moleküle, die sich gerade im Fokus befinden, an und kann daher nicht entscheiden, von welchem Molekül die Fluoreszenz gerade kommt. Daher konnten Strukturen, die kleiner sind als die Ausdehnung des Laserfokus, nicht unterschieden werden.
Anwendungen
Ein bedeutendes Problem gleich welcher lichtmikroskopischen Technik ist der mangelnde Kontrast von Zellbestandteilen. Schon lange benutzt man deshalb fluoreszente Moleküle, die z. B. mit gentechnischen Methoden oder mittels Antikörpern selektiv an bestimmte Moleküle einer Zelle geheftet werden können. Man kann zum Beispiel Farbstoffe nur an Mitochondrien anbauen. Beleuchtet man nun eine Stelle der so präparierten Zelle mit einem fokussierten Laserstrahl und erhält von dort Fluoreszenz, so waren an genau dieser Stelle Farbstoffmoleküle und damit auch Mitochondrien. Um ein vollständiges Bild zu erhalten, wird das Präparat Punkt für Punkt abgerastert. In einem STED-Mikroskop lassen sich alle Präparate untersuchen, die mit Fluoreszenzfarbstoffen markierbar sind. Anders als bei Elektronenmikroskopen sind kein Vakuum und keine dünnen Schnitte erforderlich. Deshalb lassen sich auch lebende Zellen beobachten [5].
Im Gegensatz zur Rastersondenmikroskopie ist die STED-Mikroskopie eine Fernfeld-Technik. Sie ist also nicht auf die Untersuchung von Oberflächen beschränkt. Man kann beispielsweise auch das Innere von Zellen untersuchen. Auch die Beobachtung von schnellen dynamischen Prozessen ist möglich.[6]
Leica Microsystems stellt seit 2007 kommerzielle Versionen des STED-Mikroskops her.
Literatur
- Marcus Dyba, Stefan W. Hell: Focal spots of size lambda/23 open up far-field florescence microscopy at 33 nm axial resolution. In: Physical Review Letters. Vol. 88, Nr. 16, 2002, S. 163901, doi:10.1103/PhysRevLett.88.163901.
- Katrin I. Willig, Silvio O. Rizzoli, Volker Westphal, Reinhard Jahn, Stefan W. Hell: STED-microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis. In: Nature. Vol. 440, 2005, S. 935-939, doi:10.1038/nature04592.
- Stefan W. Hell: Microscopy and its focal switch. In: Nature Methods. Vol. 6, Nr. 1, 2009, S. 24–32, doi:10.1038/nmeth.1291.
- Stefan W. Hell: Far-Field Optical Nanoscopy. In: Science. Vol. 316, 2007, S. 1153-1158, doi:10.1126/science.1137395.
Weblinks
- Abteilung NanoBiophotonics am Max-Planck-Institut für biophysikalische Chemie (englisch)
- Detailinformationen und Bilder zur STED-Mikroskopie (PDF-Datei; 426 kB)
- Hintergrundinformationen - Deutscher Zukunftspreis
- Welt der Physik - Lichtblicke in die Nanowelt
Einzelnachweise
- ↑ Stefan W. Hell and Jan Wichmann: Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. In: Optics Letters. 19, Nr. 11, 1994, S. 780–782, doi:10.1364/OL.19.000780.
- ↑ Thomas. A. Klar, Stefan W. Hell: Subdiffraction resolution in far-field fluorescence microscopy. In: Optics Letters. Vol. 24, Nr. 14, 1999, S. 954–956, doi:10.1364/OL.24.000954.
- ↑ Eva Rittweger, Kyu Young Han, Scott E. Irvine, Christian Eggeling, Stefan W. Hell: STED microscopy reveals crystal colour centres with nanometric Resolution.. In: Nature Photonics. Vol. 3, 2009, S. 144-147, doi:10.1038/nphoton.2009.2.
- ↑ Stefan W. Hell: Far-Field Optical Nanoscopy. In: Science. Vol. 316, 2007, S. 1153-1158, doi:10.1126/science.1137395.
- ↑ Volker Westphal, Silvio O. Rizzoli, Marcel A. Lauterbach, Dirk Kamin, Reinhard Jahn, Stefan W. Hell: Video-Rate Far-FieldOptical Nanoscopy Dissects Synaptic Vesicle Movement. In: Science. Vol. 320, 2008, ISSN 0036-8075, S. 246-249, doi:10.1126/science.1154228.
- ↑ Volker Westphal, Marcel A. Lauterbach, Angelo Di Nicola, Stefan W. Hell: Dynamic far-field fluorescence nanoscopy. In: New Journal of Physics. 9, 2007, S. 435ff., doi:10.1088/1367-2630/9/12/435.
Kategorien:- Lichtmikroskopie
- Bildung und Forschung in Göttingen
- Laseranwendung

Wikimedia Foundation.