- Bauchspeicheldrüsenkrebs
-
Pankreastumoren sind Tumoren der Bauchspeicheldrüse (Pankreas). Wenn in einem bildgebenden Verfahren eine Auftreibung der Bauchspeicheldrüse gefunden wird, handelt es sich in den meisten Fällen um einfache Zysten oder nach Entzündungen verbliebene Pseudozysten, die nur selten Symptome verursachen (z. B. eine Milzvenenkompression) und meist keiner Behandlung bedürfen. Etwa 30 % der bildgebend nachgewiesenen Raumforderungen in der Bauchspeicheldrüse sind jedoch echte Neoplasmen (Gewebsneubildungen). Pankreastumoren werden nach der Verwandtschaft zum normalen Gewebe und nach dem Grad der Bösartigkeit eingeteilt.
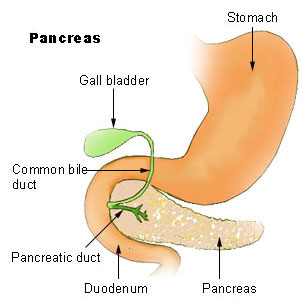
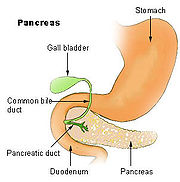 Lage des Pankreas
Lage des PankreasDie Bauchspeicheldrüse enthält exokrine Drüsenzellen, deren Verdauungssekret sich in den Azini sammelt und über die Ausführungsgänge in den Zwölffingerdarm abgegeben wird, und endokrines Gewebe, dessen Zellen Hormone produzieren und an das Blut abgeben. Beide Gewebsarten können Tumore entwickeln. Pathologen unterscheiden daher exokrine und endokrine Tumoren. 98 % der Pankreastumoren entstehen aus dem exokrinen Organ, nämlich aus dem Gangepithel und den Azinuszellen. Darunter fallen einige wenige gutartige Tumoren (Zystadenome und muzinöse Zystome); die übrigen sind bösartige Karzinome. Auch Tumoren des Binde- und Lymphgewebes, in das die Funktionsgewebe der Bauchspeicheldrüse eingebettet sind, werden zu den exokrinen Tumoren gezählt. Die Tumoren des endokrinen Pankreasgewebes (der Inselzellen) zählen zu den neuroendokrinen Tumoren des Gastrointestinaltraktes. Sie sind abgesehen von bestimmten erblichen Syndromen äußerst selten.
Drei Viertel der Tumoren entstehen im Pankreaskopf, dem am weitesten rechts, am Zwölffingerdarm gelegenen Anteil; 20 % im mittleren Anteil (Korpus) und 5 % im linksseitigen Ausläufer zur Milz hin, dem Pankreasschwanz.
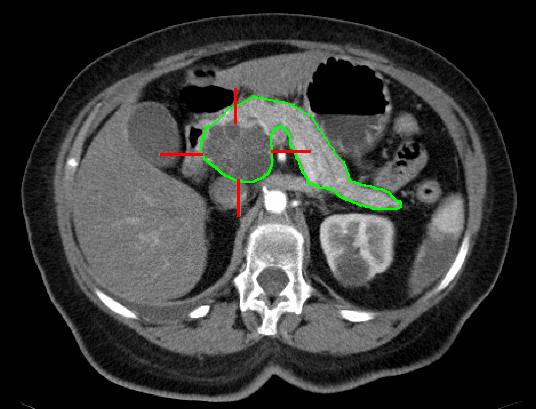
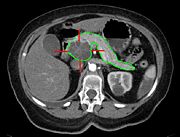 Zystisches Adenokarzinom (rot markiert) des Pankreas (grün) im CT
Zystisches Adenokarzinom (rot markiert) des Pankreas (grün) im CTInhaltsverzeichnis
Adenokarzinom
Das Pankreaskarzinom (genauer: duktales Adenokarzinom des Pankreas) mit seinen Varianten ist der bei weitem häufigste Tumor des Pankreas. Die Inzidenz liegt bei 10 Neuerkrankungen pro 100.000 Einwohner und Jahr. Es handelt sich damit um den dritthäufigsten Tumor des Verdauungstrakts. Unter den Krebserkrankungen in Deutschland steht das Pankreaskarzinom mit etwa 3 % an der 12. Stelle. Unter den Krebstodesfällen belegt es wegen seiner hohen Sterblichkeit (Letalität) in Europa je nach Land sogar den Platz 5-7.
Ursachen
Die Ätiologie des duktalen Adenokarzinoms ist unklar. Nikotinabusus, fettreiche Ernährung, chronische Pankreatitis, Diabetes mellitus, und chemische Kanzerogene (Naphthalamin, Benzidin, Nitrosamin) scheinen das Erkrankungsrisiko zu erhöhen. Genetische Untersuchungen haben Veränderungen des K-ras-Gens (Mutation am Codon 12) auf Chromosom 9p21, sowie des p53-Gens auf Chromosom 17q gefunden.
Pathologie
Der Tumor tritt bevorzugt (60 – 70 %) im Pankreaskopf auf und ist bei Diagnosestellung meist 2-5 cm groß. Er ist unscharf begrenzt, von fester Konsistenz und grau-gelblicher Farbe. Computertomografisch ist der Tumor in der arteriellen Phase hypodens demarkiert. Es kommt häufig zu einer Verengung der durch die Bauchspeicheldrüse verlaufenden Strecke des Gallengangs, häufig auch zu einer Stenose des Pankreas-Ausführungsgangs. Der Tumor kann in die Wand des Zwölffingerdarmes einwachsen, weiters auch wichtige Gefäßstrukturen, wie die A. mesenterica superior, die V. lienalis, die V. portae und/oder die V. cava inferior infiltrieren. Die Feststellung dieser Beteiligungen ist für das Staging und damit für das weitere therapeutische Procedere von größter Wichtigkeit.
_Case_01.jpg) Adenokarzinom unter dem Mikroskop
Adenokarzinom unter dem MikroskopDas histologische Bild wird geprägt durch atypische, meist noch relativ gut differenzierte gangartige Drüsenstrukturen mit Schleimproduktion. Die wichtigsten histologischen Varianten des duktalen Adenokarzinoms sind das adenosquamöse Karzinom, das muzinöse nichtzystische Karzinom und das anaplastische (undifferenzierte) Karzinom.
Die ersten Metastasen finden sich in den benachbarten Lymphknoten und – über den Blutstrom der Pfortader – in der Leber. Tumoren im Pankreaskörper und Pankreasschwanz sind bei Diagnosestellung zumeist größer als Pankreaskopftumoren und haben meistens schon zu Lebermetastasen oder einer Peritonealkarzinose (Infiltration des Bauchfells) geführt.
Symptome
Das Leitsymptom des Pankreaskopfkarzinoms ist der stetig zunehmende schmerzlose (d. h. nicht von Koliken begleitete) Ikterus, der durch die Verengung des Gallengangs verursacht wird. Im Gegensatz zum Karzinom in der Papilla Vateri (der Mündung des Gallengangs) ist der Ikterus jedoch kein Frühsymptom. Weitere häufige, aber uncharakteristische Symptome sind Gewichtsverlust und in den Rücken ausstrahlende Bauchschmerzen. Der Verschluss des Pankreasgangs führt zu einer Entzündung, die die Drüsenfunktion beeinträchtigt und zu Verdauungsbeschwerden, Gewichtsverlust und Diabetes führen kann.
Diagnostik
Die Tumoren können durch Ultraschall, Endosonographie, Kernspintomographie und Computertomographie erfasst werden. Der Verschluss des Gallen- oder Pankreasganges ist das Hauptkriterium in der ERCP. Die Tumoren produzieren meist den Tumormarker CA 19-9 und zum Teil auch das unspezifische CEA, die im Serum in erhöhter Konzentration nachgewiesen werden können.
Die TNM-Klassifikation dient zur international einheitlichen Klassifikation der Ausdehnung bösartiger Tumore. Beim Pankreaskarzinom wird sie wie folgt vorgenommen:
pT Primärtumor pTX Primärtumor kann nicht untersucht werden pT0 Kein Primärtumor nachweisbar pTis Carcinoma in situ pT1 Größter Durchmesser des Primärtumors ≤ 2 cm; Tumor innerhalb des Pankreas pT2 Größter Durchmesser des Primärtumors > 2 cm; Tumor innerhalb des Pankreas pT3 Primärtumor wächst ein in peripankreatisches Gewebe pT4 Primärtumor infiltriert angrenzende große Gefäße (Truncus coeliacus, A. mesenterica sup., V. lienalis, V. portae, V. cava inferior) pN Regionäre Lymphknoten pNX Die regionären Lymphknoten können nicht untersucht werden pN0 Keine nachweisbaren regionären Lymphknotenmetastasen pN1 Regionäre Lymphknotenmetastasen pM Fernmetastasen pMX Fernmetastasen können nicht untersucht werden pM0 Keine nachweisbaren Fernmetastasen pM1 Nachweisbare Fernmetastasen Therapie
- Die kurative Operation (Operation in Heilungsabsicht) wird oft durch die Ausbreitung des Tumors und die Ummauerung der angrenzenden Blutgefäße unmöglich gemacht. Lokal begrenzte Tumoren können chirurgisch entfernt werden.
- Palliativoperationen dienen zur Symptomlinderung: beispielsweise wird mit der Choledochojejunostomie eine Verbindung zwischen dem gestauten Gallengang und dem Darm geschaffen, um die Galle aus der Leber abzuleiten.
- Die ERCP kann außer zur Diagnose auch zur lindernden Behandlung dienen, indem ein Stent (stützendes Röhrchen) in den Gallengang eingelegt wird. Der gestaute Gallengang wird durch den Stent wieder durchgängig gemacht.
- Nicht resezierbare (nicht entfernbare) Tumoren können mit einer Chemotherapie behandelt werden, z. B. mit Gemcitabin.[1]
Verlauf und Prognose
Die Gallenstauung führt zur Funktionsstörung der Leber. Die lokale Tumorausbreitung kann außerdem eine Duodenalstenose (Einengung des Zwölffingerdarmes) und Aszites (Bauchwassersucht) bei Peritonealkarzinose zur Folge haben. Gelegentlich tritt eine Thrombophlebitis auf.
Die Prognose der Pankreaskarzinome ist schlecht. Nur 10–15 % der Tumoren sind zum Zeitpunkt der Diagnose noch operabel, und nur 1–2 % der operierten Patienten überleben fünf Jahre. Inoperable Tumoren sprechen auch schlecht auf Chemotherapie an.
Andere exokrine Pankreastumoren
Zu den Tumoren des exokrinen Pankreas zählen:
Tumortyp Häufigkeit (%) maligne Tumoren duktales Adenokarzinom mit Varianten 92 intraduktales papillär-muzinöses Karzinom 2 muzinös-zystischer Tumor (muzinöses Zystadenokarzinom) 1 Azinuszellkarzinom 1 andere Tumoren 3 benigne Tumoren seröses Zystadenom 1 Intraduktaler papillär-muzinöser Tumor (IPMT)
Synonym: Intraduktales papillär-muzinöses Karzinom. Diese Tumoren sind durch die Entstehung und Ausbreitung innerhalb des Gangsystems gekennzeichnet. Sie wachsen meist im Pankreasgang im Kopfteil der Drüse. Man unterscheidet einen Hauptgangtyp, einen Seitenasttyp und einen kombinierten Typ. Das normale Gangepithel wird durch hochzylindrische neoplastische Zellen in papillenförmigen Wucherungen ersetzt, die viskösen Schleim bilden. Letzterer fließt nur schwer ab. Dadurch kommt es zur unregelmäßigen Erweiterung des betroffenen Gangabschnitts auf 3 bis 4 cm. Die Tumorzellen können sich auch auf die Seitengänge ausbreiten und in seltenem Falle das gesamte Pankreas erfassen. Bei etwa 30 % der Patienten bestehen schon Gefäßeinbrüche und damit ein invasives Karzinom, dann meist als muzinöses nichtzystisches Karzinom (Kolloidkarzinom) bezeichnet wegen der Schleimseen im Parenchym des Organs.
Die intraduktalen papillär-muzinösen Tumoren kommen bei Männern etwas häufiger vor als bei Frauen und treten vornehmlich im Alter zwischen 60 und 70 Jahren auf. Häufige Symptome sind pankreatitisartige Schmerzen und nach langer Krankheitsdauer eine exokrine Pankreasinsuffizienz (Ausfall der Verdauungsenzyme). Die Ursache für diese Symptomatik ist die durch Schleim und Tumorgewebe bedingte Verstopfung des Pankreasgangs, die dazu führt, dass das Pankreasgewebe wie bei einer chronisch-obstruktiven Entzündung (Pankreatitis) vernarbt.
Die Prognose der intraduktal papillär-muzinösen Tumoren ist sehr gut, wenn der Tumor vollständig entfernt werden kann. Dies gilt auch für das invasive Karzinom, wenn der Tumor die Bauchspeicheldrüse noch nicht überschritten hat.
Muzinös-zystischer Tumor
Synonym: muzinöses Zystadenom, Zystadenokarzinom. Zystadenome weisen eine Entartungstendenz zum Zystadenkarzinom auf. Bildgebend kann keine Differenzierung gemacht werden, deshalb werden gut- und bösartige Varianten unter dem Begriff muzinös-zystischer Tumor zusammengefasst. Frauen im Alter zwischen 40 und 60 Jahren sind bevorzugt betroffen. Die Tumoren weisen eine breite fibröse Kapsel auf und halten etwa 2-12cm im Durchmesser. Sie bestehen in der überwiegenden Zahl aus weniger als sechs Zysten mit einem Einzeldurchmesser von >2cm. Diese Zysten sind mit muzinprduzierendem Zyliderpeithel ausgekleidet und entweder gut differenziert oder zeigen deutliche Atypien. Laborchemisch fällt ein Anstieg des Carcinoembryonalen Antigens (CEA), sowie in der überwiegenden Zahl auch des CA 19-9 auf. Gelingt die totale Resektion des Tumors, ist die Prognose gut. Bei Malignomen liegt die 5-Jahres-Überlebensrate bei etwa 75%
Azinuszellkarzinom
Dieser seltene Tumor der Azinuszellen kommt doppelt so häufig bei Männern wie bei Frauen vor (Altersgipfel: 55-65 Jahre). Obwohl die Tumoren gewöhnlich relativ groß sind (4-6 cm), werden sie oft erst entdeckt, wenn sie bereits in die Leber metastasiert haben. Gelegentlich kommt es bei Azinuszellkarzinomen, bedingt durch eine massive Sekretion von Lipasen aus den Tumorzellen, zu subkutanen Fettgewebenekrosen und Polyarthralgien.
Tumoren der Papilla Vateri
Die Tumoren im Bereich der Papilla Vateri (Papilla duodeni major, Mündung des Gallengangs) sind meist Adenokarzinome ebenso wie die duktalen Pankreaskarzinome. Gelegentlich gehen diese Karzinome aus tubulovillösen Adenomen hervor, die histologisch den Adenomen des Duodenums entsprechen. Die Prognose des Papillenkarzinoms ist besser als die des Pankreaskarzinoms, da die rasch auftretende Gelbsucht zur frühzeitigen Diagnose des Tumors führt. Ausbreitung und Metastasierung verlaufen wie beim Pankreaskarzinom.
Seröses Zystadenom
Synonym: mikrozystisches (Zyst-)Adenom. Dieser gutartige Tumor wird vorwiegend (3:2 bis 9:2) bei Frauen im höheren Lebensalter beobachtet und stellt in 10-30% einen Zufallsbefund dar. Er liegt häufiger im Pankreaskopf, jedoch kann jede Region betroffen sein. Er kann bis etwa 6-10 cm groß werden und besteht aus kleinen Zysten mit serösem Inhalt, die häufig durch zarte Septen unterteilt sind. Neben einer zentralen Narbe finden sich in mehr als einem Drittel der Fälle zentrale Verkalkungen. Die Zysten sind mit kubischem Epithel ausgekleidet, histologisch finden sich keine Atypien oder Mitosefiguren. Seröse Zystadenokarzinome sind selten. Eine Assoziation mit dem Von-Hippel-Lindau-Syndrom wurde beschrieben, der Tumor kann hier große Abschnitte des Pancreas einnehmen. Laborchemisch ist das CEA negativ. Der Tumor weist keine Entartungstendenz auf und hat nach Resektion eine gute Prognose.
Endokrine Tumoren
Zu den Tumoren des endokrinen Pankreas zählen:
- Insulinom
- Gastrinom (Zollinger-Ellison-Syndrom)
- Somatostatinom
- Glucagonom
- VIPom (Verner-Morrison-Syndrom)
Es handelt sich um Tumoren mit histologisch endokrinem Aufbau („Inselzelltumoren“). Durch die unkontrollierte Sekretion von Hormonen (Insulin, Gastrin, vasoaktives intestinales Peptid oder Glucagon) können charakteristische Syndrome hervorgerufen werden. Diese Tumoren werden daher als Insulinome, Gastrinome, VIPome und Glucagonome klassifiziert. Sog. nichtfunktionelle Tumoren weisen dagegen keine hormonelle Symptomatik auf.
Endokrine Pankreastumoren machen nur 1-2 % aller Pankreastumore aus und können in jedem Alter auftreten. Ein gehäuftes Auftreten findet man beim Syndrom der Multiple endokrine Neoplasie (MEN1-Syndrom). Ansonsten sind neuroendokrine Pankreastumoren im Kindesalter extrem selten; beim Erwachsenen treten sie in allen Altersklassen sowie bei Männern und Frauen etwa gleich häufig auf, sind insgesamt aber nicht häufig. Die Prävalenz beträgt unter 1/100000. Insulinome und Gastrinome machen 60 %, nichtfunktionelle Tumoren 30 % dieser Neoplasien aus.
Morphologie
Es handelt sich um gut begrenzte, solitäre runde Tumoren mit einem Durchmesser von 1-4 cm, die in allen Teilen des Pankreas auftreten können. Histologisch handelt es sich um monomorphe Tumorzellen mit einem feingranulären Zytoplasma. Die Zellen sind solide, trabekulär und pseudoglandulär angeordnet. Die immunzytochemische Darstellung der Hormone erlaubt eine funktionell-morphologische Einteilung dieser Tumoren. Elektronenmikroskopisch finden sich in den Tumorzellen membranbegrenzte neurosekretorische Hormongranula. Es finden sich monomorphe Zellen mit runden Zellkernen. Immunhistologisch sind endokrine Tumoren positiv für die Marker CGa, Syn und NSE.
Obwohl die neuroendokrinen Pankreastumoren histologisch hochdifferenziert sind, verhalten sie sich mit Ausnahme des Insulinoms häufig maligne. Dies gilt vor allem für Gastrinome, VIPome, Glucagonome und nichtfunktionelle Tumoren. Da die histologischen Kriterien für Malignität bei diesen Tumoren unzuverlässig sind, kann nur das Vorhandensein von Metastasen oder eine Tumorinvasion in umgebende Organe den Nachweis der Malignität sichern. Die ersten Metastasen finden sich in den regionären Lymphknoten und in der Leber. Trotz Metastasierung können häufig lange Überlebenszeiten (5-10 Jahre) beobachtet werden.
Prognose
Die Prognose bei gutartigen (benignen) Tumoren ist gut. Bei bösartigen (malignen) Formen beträgt die mittlere Überlebenszeit 1/2 Jahr, wobei auch einzelne Verläufe über 2 Jahre beschrieben sind.
Kriterien zu Einschätzung der Prognose von endokrinen Pankreastumoren (WHO 2000)
Metastase Infiltrationa Histolog Differenzierung Tumorgröße Angioinvasion Ki-67-Index Hormonelles Syndrom Benigne - - Hoch <2cm - <2% -/+ b Benigne oder niedrigmaligne - - Hoch >2cm -/+ <2% -/+ b Niedrigmaligne + + Hoch >3cm + >2% + c hochmaligne + + niedrig beliebig + >30% - - a Infiltration angrenzender Organe (z. B. Duodenum, Magen)
- b Insulinome
- c Insulinome und andere funktionell aktive Tumoren (z. B. Glucagonome)
Darüber hinaus gibt es hormoninaktive (neuro)endokrine Pankreastumore, die zwar geringe Mengen an Hormonen bilden können, diese aber nicht freisetzen und somit auch keine Symptome i.S. einer hormonellen Überproduktion verursachen (wie z. B. die o. g. Gastrinome, Insulinome etc.). Sie können lange Zeit asymptomatisch bleiben.
Ein relativ zuverlässiger Tumormarker für neuroendokrine Tumore (hormonaktiv u. hormoninaktiv) ist Chromogranin A (CgA), der im Blut bestimmt werden kann. Bei 60-80 % aller Erkrankten zeigt sich eine Erhöhung von CgA.
Statistische Informationen
In Deutschland erkranken im Jahr etwa 12.800 Personen am Pankreaskarzinom. Das durchschnittliche Erkrankungsalter der Männer liegt bei 68 Jahren, jenes der Frauen bei 75 Jahren.
Literatur und Einzelnachweise
- ↑ Burris HA, et al.: Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–2413
- W. Böcker, H. Denk, Ph. U. Heitz: Pathologie, 3. Auflage, April 2004, ISBN 3-437-42381-9
- K. J. Bühling, J. Lepenies, K. Witt: Intensivkurs Allgemein und spezielle Pathologie, 3. Auflage, 2004, ISBN 3-437-42411-4
- Syad Massalme: Crashkurs Pathologie, 1. Auflage, 2004, ISBN 3-437-43380-6
Weblinks
- Schweizer Magen-Darm-Zentrum: Bauchspeicheldrüse / Pankreas
- Netzwerk Neuroendokrine Tumoren (NeT) e.V.
- Pancreatica: Informationen rund um Behandlung, klinische Studien, Nachrichten und weiterführende Links in englisch
- EBM-Leitlinie "Exokrines Pankreaskarzinom" (S3), Dtsch. Krebsgesellschaft, Stand 10/2006 (PDF-Datei, 393 Kilobyte)
- Umfassende Informationen zur Therapie und Diagnostik des Pankreaskarzinoms der Universitätsklinik Freiburg, Stand 01/ 2009, Überblick über Studien

Bitte beachte den Hinweis zu Gesundheitsthemen!
_Case_01.jpg)
Wikimedia Foundation.