- Dopamin-β-Hydroxylase-Mangel
-
Der Dopamin-β-Hydroxylase-Mangel (DBH-Mangel), auch als Noradrenalin-Mangel bezeichnet, ist eine sehr seltene autosomal-rezessiv vererbte Stoffwechselerkrankung. Bei den betroffenen Patienten sind aufgrund eines Defektes im DBH-Gen die Hormone Adrenalin und Noradrenalin im Blutplasma nicht vorhanden, wo hingegen der Dopamin-Spiegel deutlich erhöht ist. Das Enzym Dopamin-β-Hydroxylase katalysiert die Hydroxylierung von Dopamin zu Noradrenalin, welches mit Hilfe des Enzyms Phenylethanolamin-N-Methyltransferase zu Adrenalin methyliert wird.
Das Enzym Dopamin-β-Hydroxylase katalysiert die Hydroxylierung von Dopamin zu Noradrenalin, welches mit Hilfe des Enzyms Phenylethanolamin-N-Methyltransferase zu Adrenalin methyliert wird.
Inhaltsverzeichnis
Prävalenz und Symptome
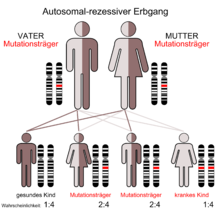 Autosomal-rezessiver Erbgang
Autosomal-rezessiver ErbgangDie genaue Prävalenz des Dopamin-β-Hydroxylase-Mangels ist nicht bekannt, liegt aber offensichtlich bei einem Wert kleiner als 1 : 1 Million. Die Erkrankung ist äußerst selten und es wurden bisher nur sehr wenige Patienten beschrieben.[1] Die Prävalenz der Erkrankung ist vermutlich deutlich höher, da die Erkrankung zu Fehlgeburten führen kann. Im Tiermodell Maus ist der Dopamin-β-Hydroxylase-Mangel häufig schon pränatal tödlich.[2][1]
Kardiovaskuläre Störungen und eine schwere orthostatische Hypotonie sind die wesentlichen Symptome bei Dopamin-β-Hydroxylase-Mangel und treten schon unmittelbar nach der Geburt auf. Niedriger Blutdruck, Muskelhypotonie, Hypothermie und Hypoglykämie sind die in der Neugeborenenperiode auftretenden Komplikationen. Da sich der Blutdruck wechselnden körperlichen Belastungen nicht anpassen kann, vertragen die betroffenen Kinder keine aktive Bewegung oder körperliche Anstrengungen. Dieser Zustand verschlechtert sich mit zunehmendem Alter weiter. Im frühen Erwachsenenalter sind daher unter anderem schwere orthostatische Hypotension und Ptosis zu beobachten. Stehen über einen längeren Zeitraum ist für die Betroffenen nicht möglich. Kreislaufkollapse, die Sturzverletzungen zur Folge haben, sind häufig.[1]
Genetik und Pathogenese
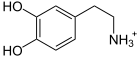 + Ascorbat + O2 →
+ Ascorbat + O2 → 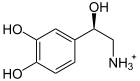 + Dehydroascorbat + H2O
+ Dehydroascorbat + H2ODas für das Enzym Dopamin-β-Hydroxylase kodierende DBH-Gen befindet sich beim Menschen auf Chromosom 9 Genlocus q34.[3] Das DBH-Gen ist ungefähr 23 kb lang und besteht aus 12 Exons.[4] Mutationen im DBH-Gen können bewirken, dass das Genprodukt – die Dopamin-β-Hydroxylase – in seiner Funktion eingeschränkt oder gar vollständig defekt ist. Das Enzym katalysiert die Oxidation von Dopamin zu Noradrenalin. Ist die Aktivität durch Mutationen im DBH-Gen eingeschränkt, so wird kaum oder kein Noradrenalin und in der weiteren Abfolge kein Adrenalin aus Dopamin produziert. Auf der anderen Seite reichert sich der Ausgangsstoff Dopamin im Plasma an.
Die Mutationen im DBH-Gen werden autosomal-rezessiv vererbt.[5] Bisher wurden vor allem Missense-Mutationen auf den Exons 1, 2 oder 6 festgestellt.[1]
Diagnose
Dopamin-β-Hydroxylase ist bei etwa 4% der Bevölkerung mit normalen Katecholamin-Konzentrationen nicht nachweisbar[6], so dass die Bestimmung des Enzyms alleine keine sichere Diagnose darstellt. Der erhöhte Plasmaspiegel von Dopamin und die Abwesenheit von messbaren Mengen an Noradrenalin und Adrenalin im Blut der Patienten ermöglicht eine sichere Labordiagnose von Dopamin-β-Hydroxylase-Mangel.[7] Im Urin fehlen zudem die Stoffwechselprodukte von Noradrenalin.[1]
Therapie
 Droxidopa, ein Prekursor für Noradrenalin
Droxidopa, ein Prekursor für NoradrenalinDer Dopamin-β-Hydroxylase-Mangel kann durch die Gabe von Droxidopa (L-threo-3,4-Dihyroxyphenylserin) behandelt werden.[8] Droxidopa ist ein Vorläufermolekül (Prodrug) von Noradrenalin.[9][10]
Im Tiermodell Maus mit abgeschaltetem Dhb-Gen (Knockout-Maus) bewirkt die Gabe von Droxidopa, dass die Plasmakonzentration an Noradrenalin normale Werte annimmt und die Verhaltensstörungen der Mäuse verschwinden.[11] Beim erkrankten Menschen bewirkt die gleiche Substanz eine drastische Erhöhung des Blutdruckes und eine Linderung der posturalen Symptome.[12][1]
Der Wirkstoff wird üblicherweilse zwei oder dreimal täglich oral eingenommen und korrigiert die Gleichgewichtsstörungen.[1]
Prognose
Über die Prognose der Erkrankung sind bisher nur sehr wenige Daten vorhanden. Die Gabe von Droxidopa ist eine sehr effektive Therapieform, um die wichtigsten Symptome des Dopamin-β-Hydroxylase-Mangel deutlich zu lindern.[1]
Erstbeschreibung
Der Dopamin-β-Hydroxylase-Mangel wurde erstmals 1986 von einer Arbeitsgruppe um den Arzt David Robertson an der Vanderbilt University in Nashville beschrieben.[13]
Weiterführende Literatur
- J. F. Cubells und C. P. Zabetian: Human genetics of plasma dopamine beta-hydroxylase activity: applications to research in psychiatry and neurology. In: Psychopharmacology (Berl) 174, 2004, S. 463–476. PMID 15088079 (Review)
- H. J. Timmers u. a.: Congenital dopamine-beta-hydroxylase deficiency in humans. In: Ann N Y Acad Sci 1018, 2004, S. 520–523. PMID 15240410 (Review)
- D. Robertson u. a.: Dopamine-b-hydroxylase deficiency and cardiovascular control. In: Hypertension Pathophysiology, Diagnosis and Management. Raven Press Ltd, New York, 1990, S. 749–759.
Weblinks
- Dopamin-β-Hydroxylase-Mangel bei Online Mendelian Inheritance in Man
- Dopamin-β-Hydroxylase-Mangel bei Orphanet (Datenbank für seltene Krankheiten)
- Dopamine beta-hydroxylase deficiency (englisch)
- Dopamine beta-hydroxylase deficiency (englisch)
Einzelnachweise
- ↑ a b c d e f g h J. M. Senard und P. Rouet: Dopamine beta-hydroxylase deficiency. In: Orphanet J Rare Dis 1, 2006, 7 PMID 16722595 (Review, Open Access unter CC-by-2.0)
- ↑ S. A. Thomas: Noradrenaline is essential for mouse foetal development. In: Nature 374, 1995, S. 643–646. PMID 7715704
- ↑ S. P. Craig u. a.: Localization of the human dopamine beta hydroxylase (DBH) gene to chromosome 9q34. In: Cytogenet Cell Genet 48, 1988, S. 48–50. PMID 3180847
- ↑ K. Kobayashi u. a.: Human dopamine beta-hydroxylase gene: two mRNA types having different 3'-terminal regions are produced through alternative polyadenylation. In: Nucleic Acids Res 17, 1989, S. 1089–1102. PMID 2922261
- ↑ C. H. Kim u. a.: Mutations in the dopamine β-hydroxylase gene are associated with human norepinephrine deficiency. In: Am J Med Genet 108, 2002, S. 140–147. PMID 11857564
- ↑ R. M. Weinshilboum u. a.: Inheritance of very low serum dopamine-betahydroxylase activity. In: Am J Hum Genet 27, 1975, S. 573–585. PMID 1163533
- ↑ D. Robertson u. a.: Dopamine β-hydroxylase deficiency. A genetic disorder of cardiovascular regulation. In: Hypertension 18, 1991, S. 1–8. PMID 1677640
- ↑ S. Braune und C. H. Lücking: Orthostatische Hypotonie: Pathophysiologie, Differentialdiagnose und Therapie. In: Dtsch Arztebl 94, 1997, S. A-3413/B-2877/C-2673
- ↑ D. S. Goldstein: L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug. In: Cardiovasc Drug Rev 24, 2006, S. 189–203. PMID 17214596 (Review)
- ↑ C. J. Mathias: L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension: the European experience. In: Clin Auton Res 18, 2008, S. 25–29. PMID 18368304 (Review)
- ↑ S. A. Thomas u. a.: Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine β-hydroxylase. In: J Neurochem 70, 1998, S. 2468–2476. PMID 9603211
- ↑ I. Biaggioni und D. Robertson: Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. In: The Lancet 330, 1987, S. 1170–1172. PMID 2890806
- ↑ D. Robertson u. a.: Isolated failure of autonomic noradrenergic neurotransmission. Evidence for impaired β-hydroxylation of dopamine. In: NEJM 314, 1986, S. 1494–1497. PMID 3010116

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in der Kinderheilkunde
- Krankheitsbild in der Neurologie
- Stoffwechselkrankheit
- Erbkrankheit




Wikimedia Foundation.