- Hemmkörper-Hämophilie
-
Klassifikation nach ICD-10 D66 Hereditärer Faktor-VIII-Mangel D67 Hereditärer Faktor-IX-Mangel D68.- Sonstige Koagulopathien ICD-10 online (WHO-Version 2006) Hämophilie (altgriech. αἷμα haima „Blut“, gr. φίλος philos „Freund“; auch Bluterkrankheit) ist eine Erbkrankheit, bei der die Blutgerinnung gestört ist. Das Blut aus Wunden gerinnt nicht oder nur langsam. Häufig kommt es auch zu spontanen Blutungen, die ohne sichtbare Wunden auftreten. Hämophilie tritt hauptsächlich bei Männern auf.
Inhaltsverzeichnis
Formen
Es gibt im engeren Sinne zwei bekannte Formen der Hämophilie sowie weitere Krankheitsbilder, die gelegentlich unscharf unter diesem Begriff subsumiert werden:
- Hämophilie A (X-chromosomal-rezessiv erblicher Gerinnungsdefekt): Hiervon sind nahezu ausnahmslos (bis auf weltweit zwei dokumentierte Fälle) Männer betroffen, da diese nur ein X-Geschlechtschromosom besitzen, während Frauen davon zwei besitzen. Hier kommt es zu einem Mangel an Faktor VIII (antihämophiles Globulin).
- Hämophilie B (ebenfalls X-chromosomal-rezessive Vererbung): Mangel an Faktor IX (Christmas-Faktor) der Gerinnungskaskade mit verschiedenen Verläufen von Geburt an (schwer, mittelschwer, leicht). Durch diesen Mangel kann die Blutgerinnung nur sehr langsam verlaufen.
- Der sehr seltene autosomal-rezessiv erbliche Gerinnungsdefekt (z. B. Stuart-Prower-Faktor-Mangel, Faktor X der Gerinnungskaskade) kann sich bei beiden Geschlechtern gleich stark ausprägen, da bei beiden Geschlechtern gleich viele Autosomen (nicht-geschlechtsgebundene Chromosomen) vorkommen.
- Parahämophilie (Hypoproakzelerinämie, Owren-Syndrom): autosomal-rezessiv erbliche Krankheit durch Mangel des Gerinnungsfaktors V (Proaccelerin).
- Angiohämophilie (besser: Willebrand-Jürgens-Syndrom): Es ist die häufigste autosomal-dominant vererbte Gerinnungsstörung, sie entsteht durch einen Strukturdefekt oder einen Mangel des von-Willebrand-Faktors unterschiedlicher Ausprägung. Der von-Willebrand-Faktor ist ein Trägerprotein des Blutgerinnungsfaktors VIII.
- Hämophilie C (Rosenthal-Syndrom): Hier fehlt Faktor XI (PTA) der Gerinnungskaskade, so dass vor allem bei Kindern leicht Blutungen in Gelenken oder bei minimalen Verletzungen auftreten. Wie bei Hämophilie A und B ist der Quickwert typischerweise normal, während die PTT abhängig von der Ausprägung des Mangels verlängert ist.
Symptome
Hämophilie-Patienten bluten dauerhafter als Gesunde. Je nach Schweregrad können sogar Spontanblutungen auftreten, d. h. Blutungen ohne entsprechende Verletzung. Diese Spontanblutungen treten bei Gesunden ebenfalls und genauso häufig auf, heilen jedoch rasch und unbemerkt. Die Blutungen können im Prinzip überall auftreten, jedoch sind bestimmte Lokalisationen typisch für Blutungen bei Hämophilie z. B. Gelenkeinblutungen s. u.
Eine durch Unfall hervorgerufene oder eine schwere Blutung kann nur durch Gabe von Gerinnungsfaktoren in Grenzen gehalten werden. Ist diese Hilfe nicht rechtzeitig möglich, kann das (auch bei weniger schweren Verletzungen) den Tod durch Verbluten bedeuten.
Entgegen einer weit verbreiteten Vorstellung und immer noch viel zitierten alten Fachbuchartikeln führen Schnitt-, Riss- und Schürfwunden bei den häufigsten Unterformen der Hämophilie zunächst nicht zu stärkerem Blutverlust als bei gesunden Menschen, da die Krustenbildung dank der intakten Blutplättchen (Thrombozyten) zunächst funktioniert. Erst die verzögerte Blutgerinnung führt dazu, dass die Verkrustung immer wieder aufbrechen kann und die Blutung je nach Schweregrad der Hämophilie nur sehr langsam oder gar nicht zum Stehen kommt. Auch ohne äußere Einwirkung kann es zu subkutanen oder intramuskulären Hämatomen kommen.
Die Gefahr innerer Blutungen ist ebenfalls höher als normal einzustufen - (z. B. Nierenblutungen mit starker Kolik (Verschluss der Harnwege durch Thromben)).
Bei Trägerinnen des Gendefekts (sog. Konduktorinnen) kann eine verstärkte Blutungsneigung auftreten, die sich in verstärkten Regelblutungen, Neigung zu blauen Flecken (Hämatomen), bei Bagatelleingriffen wie Zahnextraktionen oder während bzw. nach Entbindung zeigen kann. In seltenen Fällen und dann meist bei ausgeprägtem Gerinnungsdefekt können auch Blutungen auftreten, die denen von männlichen Betroffenen gleichen (z. B. Gelenkblutungen).
Gelenkblutungen und ihre Folgen
Eine häufige Lokalisation sind Gelenkblutungen (Hämarthros). Die erste Blutung in einem Gelenk (auch als Initialblutung bezeichnet) wird häufig durch einen Unfall/Trauma verursacht. Besonders betroffen sind die großen Gelenke. Durch die Gelenkinnenhaut (Synovia) werden Enzyme freigesetzt, die das im Gelenk befindliche Blut abbauen. Bei großvolumigen Ergüssen vergrößert sich die Synovia dafür und wird stärker mit Blutgefäßen durchzogen. Daraus folgt eine höhere Wahrscheinlichkeit nachfolgender Blutungen oder Entzündungen. Es wird ein Kreislauf von Entzündungen und Blutungen in Gang gesetzt, es entsteht eine so genannte Hämarthrose; insbesondere ungeführte Bewegungen, insbesondere Torsionen und Überstreckungen (auch in der Nacht) „umknicken“, stolpern etc. können weitere Gelenkblutungen (meist Sprunggelenk-, Knie-, Ellenbogen-, Schulter- oder selten Hüftblutungen) zur Folge haben, was immer auch mit starken Schmerzen verbunden ist. Die Folgen der häufigen Blutungsereignisse sind bei älteren Jahrgängen (da wirksame prophylaktische Therapien erst seit etwa 30 Jahren verfügbar sind) Gelenkversteifungen z. T. schwerster Art, frühzeitige Arthrose – (die evtl. operative Eingriffe wie z. B. Knie-Arthroskopie, Synovektomie bis hin zur Endoprothese (Gelenkersatz) aber auch orthopädische Hilfsmittel (orthopädische Schuhe), Gehhilfen u. a.) erforderlich machen, – sowie Fehlbildungen der Muskulatur und des Knochenaufbaus, wobei die Mobilität der Gelenke durch ständige Physiotherapie auf einem gewissen Belastungsgrad gehalten, oder aber auch verbessert werden kann.
Muskelblutungen und ihre Folgen
Muskelblutungen treten weniger oft spontan auf als Gelenkblutungen und haben meistens Traumata als Ursache. Je nach Lage und Größe des Muskels können sie jedoch extrem langwierig werden und durch irreversible Muskelschädigung zu Verkrüppelungen führen. Muskelblutungen können auch nach Impfungen auftreten, die üblicherweise in einen Muskel (z. B. Gesäßmuskel, Oberarmmuskel) gegeben werden. Bluterpatienten sollten daher Impfungen nur unter die Haut erhalten („subkutan“). Typische gefährliche Muskelblutungen sind zu finden im bzw. bei den:
- Psoasmuskel (verläuft vom Bauch durch das Becken zum Bein, so dass ein „Stillhalten“ während des Heilungsprozesses fast nicht möglich ist und die Blutung oft nach scheinbarer Heilung wiederkehrt). Die dadurch entstehenden Schmerzen wurden besonders vor Einführung von Ultraschalluntersuchungen mit einer Blinddarmentzündung verwechselt (sog. „Pseudoappendizitis“)
- Wadenmuskel (Wadenblutungen führen zu einer Verkürzung des Muskels und dadurch zum Spitzfuß, der wiederum zu einer erhöhten Belastung der Wade beim Gehen führt und zu weiteren Blutungen führen kann)
- Unterarme (Blutungen der Unterarme können auf die Handnerven drücken und neben extremen Schmerzen auch Unbeweglichkeiten und Fehlstellungen der Hände auslösen)
Therapie
Behandlung mit dem fehlenden Faktor VIII (Hämophilie A) oder Faktor IX (Hämophilie B)
Die frühere (bis etwa 1970) gebräuchliche Therapie bei Hämophilie Blutungen zu stoppen, bestand im Allgemeinen darin, direkte Blutspende, Blutkonserven oder Blutplasma bei stärkeren und akuten Blutungen zu verabreichen, Hämatome zu kühlen, und blutende Wunden mit aus Rinderblut gewonnenem Fibrin zum Gerinnen zu bringen, was relativ selten gelang.
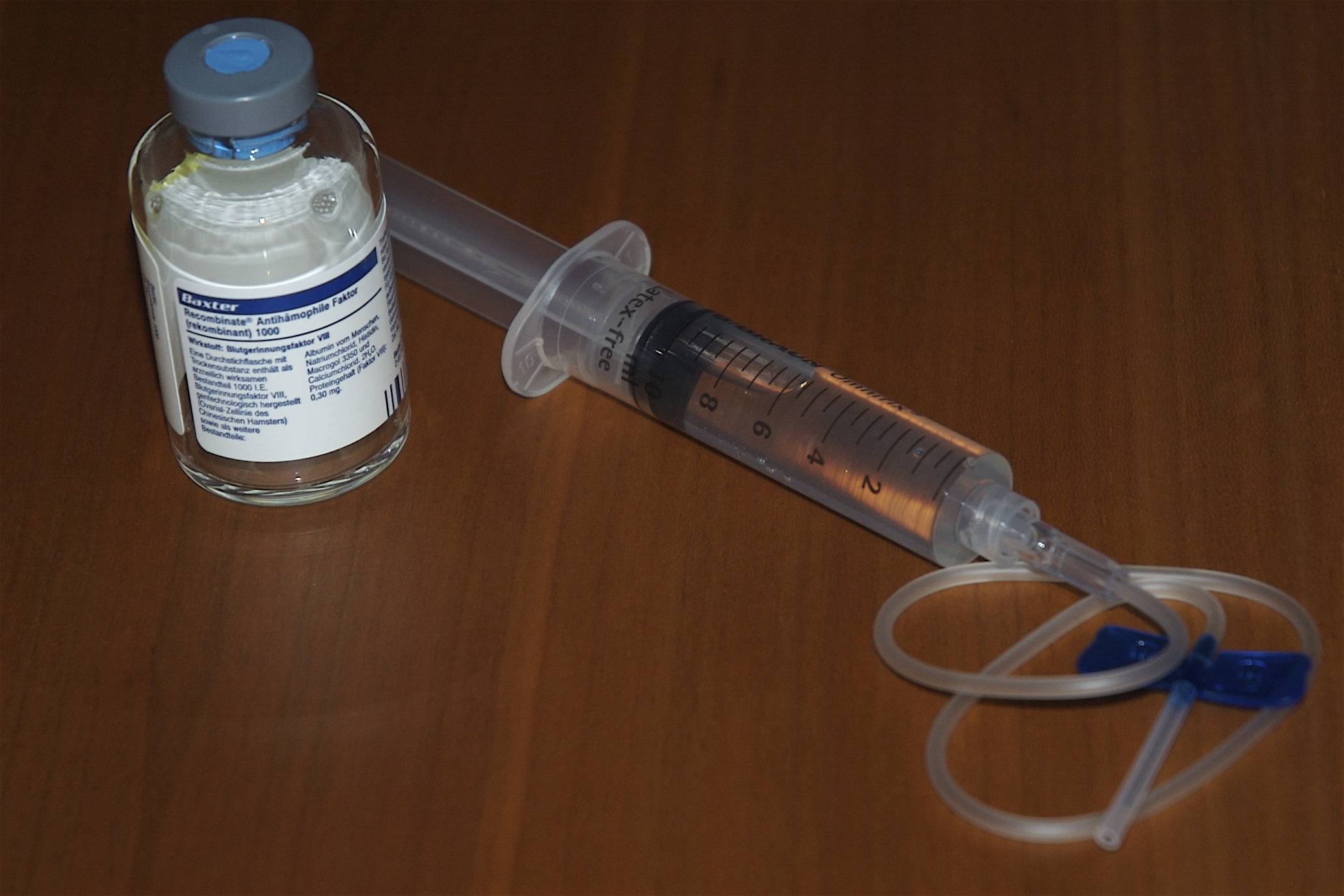
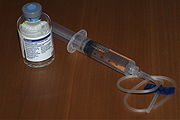 Selbstbehandlung
SelbstbehandlungDie heutige Therapie besteht im allgemeinen darin, prophylaktisch oder bei Bedarf den fehlenden oder defekten Faktor zu substituieren, wobei Blutungen weitestgehend ausgeschlossen werden können, und der Patient ein relativ normales Leben führen kann, aber z. B. von Sportarten wie Athletik, Boxen, Wintersport und extremer körperlicher Belastung absehen muss. Die Therapie erfolgt z. B. in den Fällen Hämophilie A, B oder Willebrandt-Syndrom durch Selbstbehandlung (intravenös) mit den fehlenden Faktoren. Diese Faktoren wurden bis ca. 2002 meistens aus menschlichem Blutplasma gewonnen, wobei in der Vergangenheit u. a. auch viele Bluter mit HIV, Hepatitis C und B und anderen Viren infiziert wurden. Dies wurde als sogenannter "Blutskandal" bekannt. Die Möglichkeit der Ansteckung kann seit ca. 1988 (das Hepatitis-C-Virus wurde erst Ende der 80er Jahre entdeckt) jedoch so gut wie ausgeschlossen werden, wenn die existierenden Methoden der Blutreinigung und Virusinaktivierung bestimmungsgemäß angewendet werden. Seit ca. 1989 wird der Faktor VIII (Hämophilie A) auch gentechnisch hergestellt. (siehe auch Gentechnik innerhalb dieses Absatzes), um eine intrinsische Sicherheit vor Verunreinigungen des Faktor VIII z. B. mit Viren zu bieten und um jederzeit eine ausreichende Versorgung der Patienten sicherzustellen. Diese gentechnologisch hergestellten Konzentrate gelten heute als die sichersten. Das Faktor IX Konzentrat muss noch aus Human-Blut hergestellt werden. Das gentechnische Präparat befindet sich noch in der Entwicklungsphase.
Komplikation
Die Hauptkomplikation bei der Hämophilie A-Therapie liegt heute in der Bildung von neutralisierenden Antikörpern gegen den Faktor VIII (FVIII), den sogenannten inhibitorischen Antikörpern oder auch Hemmkörpern. Diese Komplikation wird auch als Hemmkörperhämophilie oder Immunhemmkörperhämophilie bezeichnet. Weltweite Studien zeigen, dass etwa 30 % der behandelten Patienten oder Blutern inhibitorische Antikörper entwickeln. Es wird weiterhin diskutiert, ob die Inhibition allein durch die Blockierung der FVIII-Aktivität erfolgt, oder ob es zu einer erhöhten Beseitigung (engl.: clearance) des FVIII durch die Erkennung der Antikörper kommt. Die Antikörper verringern die Wirkung des zugegebenen FVIII sehr stark, so dass die nötige Erhöhung des Faktorspiegels nicht erreicht wird, und es in der Folge wieder zu Blutungen kommt.
Eine Hemmkörperhämophilie kann auch bei Substitution von Faktor IX, das heißt bei der Therapie der Hämophilie B auftreten. Sie kommt jedoch deutlich seltener vor – in 2 bis 5 Prozent der Fälle.[1]
Vererbung
Die Erkrankung wird X-chromosomal-rezessiv vererbt. Frauen können Trägerinnen für die Vererbung der Hämophilie A oder B sein, ohne selbst an der Krankheit zu leiden. Beispiel: Eine Trägerin (Konduktorin) des fehlerhaften Gens für die Hämophilie, bei der das Merkmal nicht ausgeprägt ist, bekommt Söhne, bei denen die Wahrscheinlichkeit 50 % ist, Bluter zu sein (siehe auch Erbinformation). Bekommt diese Trägerin Töchter, können statistisch 50 % dieser das Gen auf die nächste Generation weitervererben, ohne selbst von dieser Krankheit betroffen zu sein. Sobald diese Mädchen wieder männliche Nachkommen haben, ist es dann ebenso möglich, dass diese Bluter sind. Aufgrund dieser Wahrscheinlichkeit kann aber die Krankheit auch mehrere Generationen überspringen, sofern immer wieder Töchter als Trägerinnen vorhanden waren. Wenn Bluter Söhne bekommen, vererben sie die Krankheit an diese nicht weiter, da sie X-Chromosomal vererbt wird. Männliche Bluter können die Krankheit somit nur an ihre Töchter vererben.
In Einzelfällen ist die Hämophilie A bzw. B bei Frauen möglich. Wenn der Vater Bluter und die Mutter Überträgerin ist und die Tochter von der Mutter das merkmalstragende X-Chromosom vererbt bekommt (50 prozentige Wahrscheinlichkeit), wird die Tochter Bluter sein. Es gibt jedoch nur einzelne, meist sehr schlecht dokumentierte Fälle von Hämophilie bei Frauen. Die gelegentliche Erwähnung von hämophilen Frauen in Literatur und Belletristik wird mit einer Fehlzuschreibung anderer Gerinnungsstörungen erklärt.
Gentechnik
Bei Hämophilie A und B wurde das defekte Gen vor kurzem entschlüsselt und es besteht in Zukunft evtl. die Möglichkeit dieses zu reparieren und wieder funktionstüchtig zu machen, wobei die Gabe der fehlenden Faktoren in Zukunft entfallen würde und die Betroffenen ein ganz normales Leben mit normaler Gerinnung führen können.
Die Gentechnik (ein Teilgebiet der Biotechnologie) brachte in Bezug auf die Herstellung von Faktor-VIII-Präparaten bahnbrechende Fortschritte. Die Faktor-VIII-Präparate wurden bis vor einigen Jahren aus dem Blutplasma von – vermeintlich – Gesunden, also Nichtblutern, gewonnen. Durch die Verunreinigungen des Blutplasma mit – bis dato – unbekannten Viren (HIV und Hepatitis-C-Virus) kam es in den achtziger Jahren zu dramatischen Infektionen von Hämophilen. Zwischenzeitlich kann man davon ausgehen, dass bekannte Viren in plasmatischen Präparaten herausgefiltert oder abgetötet werden. Zweifellos besteht zwangsläufig ein Restrisiko hinsichtlich neuer, unbekannter Viren.
Großbritannien z. B. hat aus den Blutskandalen der vorigen Jahrzehnte gelernt – nicht zuletzt ist England auch mit der Creutzfeldt-Jakob-Krankheit entsprechend geschlagen – und plasmatische Präparate vollkommen verboten. Denn es ist mittlerweile möglich, den Blutgerinnungsfaktor VIII gentechnisch herzustellen. Er wird maschinell in Zellkulturen von Hamstern synthetisiert, isoliert und gereinigt. Das daraus entstandene Präparat wird Hämophilen (Bluterkranken) gespritzt, um Blutungen zu stoppen.
Seit einigen Jahren gibt es jetzt Faktor-VIII-Präparate, die auf biotechnologischer Basis hergestellt werden. Und selbst hier gibt es noch Unterschiede, ob im Herstellungsprozess noch Blutplasma und Albumin verwendet wird oder ob die Zellen quasi „Vegetarier“ sind und daher ganz ohne Zusatz von menschlichen oder tierischen Plasmaproteinen auskommen. Die letzt genannten können weder bekannte noch unbekannte oder in der Zukunft auftretende Viren oder Bakterien übertragen. Auf jeden Fall bieten die biotechnisch hergestellten Präparate eine sehr viel höhere Sicherheit vor Viren und anderen Verunreinigungen als direkt aus Blutplasma hergestellte Faktor VIII-Präparate (siehe auch Octocog-alfa)..
Geschichte
Die wahrscheinlich früheste Erwähnung der Krankheit findet sich im 5. Jahrhundert im Talmud, der von der rituellen Beschneidung derjenigen Knaben befreit, deren zwei Brüder bei der Beschneidung gestorben seien. In der Vergangenheit litten viele Adlige und Mitglieder der Königsfamilien an Hämophilie, weshalb sie auch den Namen „Krankheit der Könige“ erhielt. Bekannte Beispiele dafür sind die britische Königs- und die russische Zarenfamilie. Ausgangspunkt war hier die Trägerin der Krankheit Queen Victoria von England, deren Enkelin Alix von Hessen-Darmstadt den Zaren Nikolaus II. heiratete und die Krankheit auf ihren gemeinsamen Sohn Alexei übertrug.
Literatur
- Mario von Depka Prondzinski, Karin Kurnik: Hämophilie. Ein Leitfaden für Patienten. Trias-Verlag Stuttgart, 2008, ISBN 978-3-8304-3432-0
- Louis Hovy, Karin Kurnik, Mario von Depka: Hämophilie und Orthopädie. Ein interaktives Lehrbuch. Thieme, Stuttgart/New York 2004, ISBN 3-13-132981-5 (CD-ROM; ab Pentium III, Windows 98, Audiofunktionalität)
- Mario von Depka: Blutgerinnung. Aktuelle Aspekte der Physiologie, Pathophysiologie, Klinik, Diagnostik, Prophylaxe und Therapie. UNI-MED, Bremen/London/Boston 2002, ISBN 3-89599-554-1
Einzelnachweise
- ↑ H. Renz-Polster, S. Krautzig: Basislehrbuch Innere Medizin. Urban & Fischer-Verlag München 2008, 4. Auflage, S. 342 ff. ISBN 978-3-437-41053-6
Weblinks
- Österreichische Hämophilie Gesellschaft
- Schweizer Hämophiliegesellschaft
- Deutsche Hämophiliegesellschaft
- Interessengemeinschaft Hämophiler e. V. (Patientenverband für das Krankheitsbild Hämophilie)
- Bluter-Info (Liste der Hämophilie-Ambulanzen in Deutschland)
- Hämophilie – Patienteninfo von NetDoktor.at
- haemophilie.org

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.