- Alpha-Helix
-
Die α-Helix ist ein häufig auftretendes Motiv der Sekundärstruktur eines Polypeptids bzw. Proteins. Andere Motive sind das β-Faltblatt und drei verschiedene Arten von β-Schleifen. Als Sekundärstruktur bezeichnet man die räumliche Lage der Aminosäurenketten ohne Berücksichtigung der Seitengruppen (allgemeine Bezeichnung: „R“). Die nicht zu einem Motiv gehörenden Teile der Aminosäureketten eines Proteins nennt man Random-Coil-Strukturen.
Proteine, die innerhalb eines Lebewesens durch den Körper im Zuge der Biosynthese aus den elementaren Bestandteilen wie Aminosäuren und Spurenelementen hergestellt werden, erfüllen in jedem Lebewesen eine bestimmte Funktion. So gibt es Enzyme, die uns die Verdauung ermöglichen, indem sie z. B. durch die Nahrung aufgenommene Proteine (Eiweiße) spalten und in resorbierbare Stücke zerteilen, oder Antikörper, die uns gegen Infekte schützen. Das bedeutet, dass jedes Protein, das vom Körper hergestellt wird, einem definierten Zweck dient, und dementsprechend selektiv sein muss. Diese Selektivität wird häufig durch entsprechende räumliche Kompatibilität des Proteins und dessen Ziel (Substrat) sichergestellt. Dadurch wird sichergestellt, dass die Vorgänge innerhalb des Lebewesens geordnet ablaufen, ja, überhaupt erst funktionieren können. Um diese Stringenz hinsichtlich der Proteine und deren Substrate zu erreichen, haben sich im Laufe der Evolution einige sogenannte Sekundärstrukturen etabliert. Das bedeutet, dass Proteine, die sich aus einer definierten Reihenfolge von Aminosäuren, der sogenannten Primärstruktur oder auch Aminosäuresequenz, zusammensetzen, sich zu Motiven zusammenfalten, die man immer wieder bei der Analyse von Proteinen unterschiedlichster Herkunft beobachten kann (z. B. α-Helix, β-Faltblatt). Diese Sekundärstrukturen setzen sich dann innerhalb des Proteins, verbunden durch Peptidketten unterschiedlichster Konformation, zu einer Art funktionellen Gruppen zusammen und bilden die räumlich koordinierte Tertiärstruktur. Mit Hilfe dieser Struktur wird die gewünschte Selektivität erzielt i.e. die Proteinaktivität auf ein Substrat oder eine Gruppe von Substraten beschränkt (Motive innerhalb der Tertiärstruktur: z. B. HLH-Motiv, HTH-Motiv, TIM-Barrel, Zink-Finger oder Rossmann-Fold).
Der α-Helix fällt dabei eine interessante Bedeutung zu, da sie zu den stabilsten natürlichen Konformationen einer Peptidsequenz gehört und allgegenwärtig (ubiquitär) ist.
Inhaltsverzeichnis
Struktur und Funktion
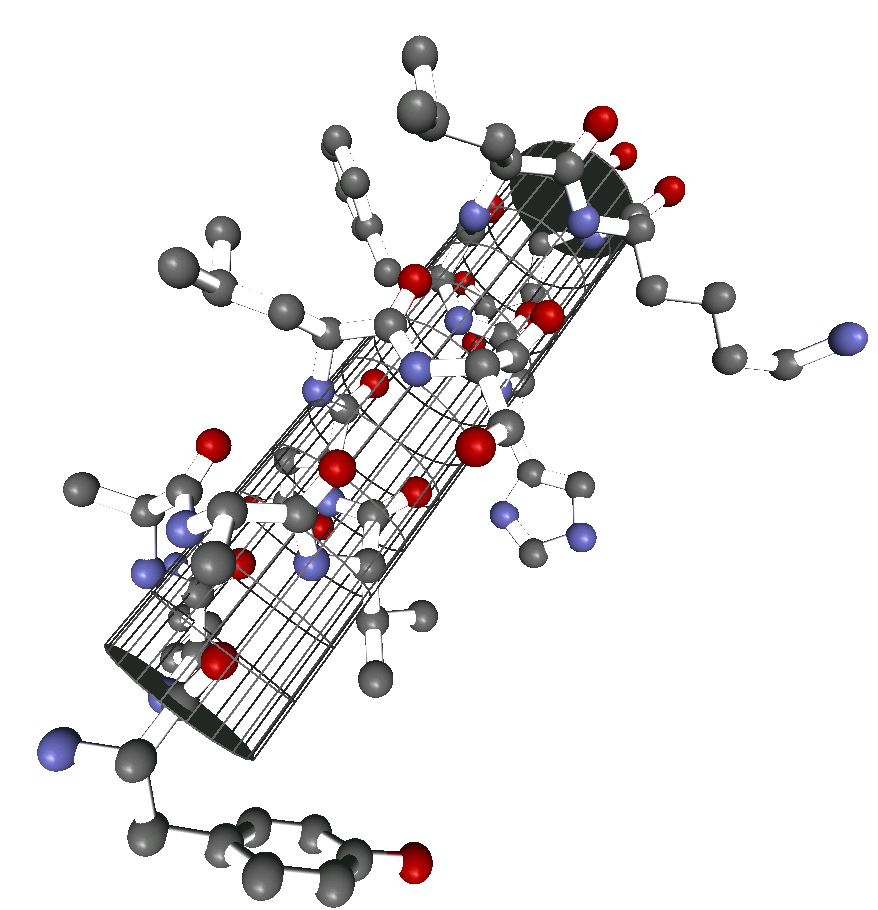
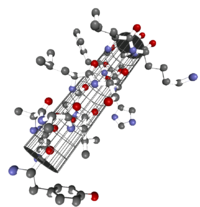 α-Helix innerhalb eines Proteins. Darstellung der Atome und Zylinder-Darstellung.
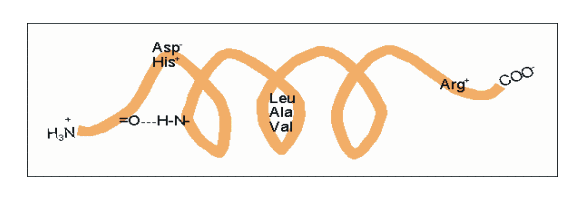
α-Helix innerhalb eines Proteins. Darstellung der Atome und Zylinder-Darstellung.Die α-Helix ist eine rechtshändig gedrehte Spirale mit durchschnittlich 3,6 Aminosäureseitenketten pro Umdrehung. Pro Windung wird eine Länge von p = 0,54 nm (5,4 Å) erzielt. Diesen Fortschritt bezeichnet man auch als Ganghöhe. Sie ist das Produkt aus Schiebung (auch Translation genannt) (0,15 nm) und Resten pro Windung (3,6). Dieser Abstand zwischen den Resten ist der Grund dafür, dass Aminosäuren, die in der Primärstruktur drei oder vier Reste voneinander entfernt sind, sich in der Helixstruktur in unmittelbarer Nähe befinden. Stabilisiert wird sie durch eine Wasserstoffbrückenbindung zwischen dem Carbonylsauerstoff der n-ten und dem Amidproton der (n+4)-ten Aminosäure desselben Moleküls.
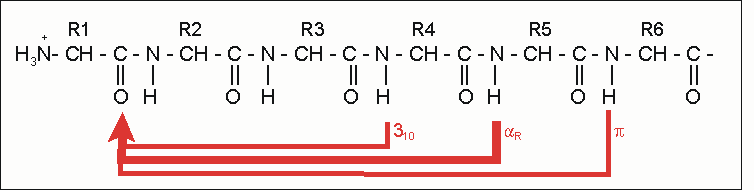
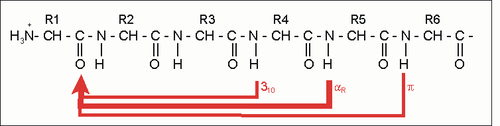 Stabilisierungsschema bei Protein-Helices: Die häufigste und stabilste Helix ist die α-Helix (dicker roter Pfeil). Alternativen existieren, sind aber seltener (dünne Linien).
Stabilisierungsschema bei Protein-Helices: Die häufigste und stabilste Helix ist die α-Helix (dicker roter Pfeil). Alternativen existieren, sind aber seltener (dünne Linien).Die CO- und NH-Gruppen müssen zur Ausbildung der Wasserstoffbrückenbindung dicht beieinander liegen. Die engste Konfiguration liefert dabei ein aufgewickelter Strang, bei der die beiden Gruppen übereinander zu liegen kommen. Die Seitenketten zeigen dabei nach außen. Die Aminosäure Prolin („Strukturbrecher“) lässt sich nicht ohne weiteres in die Helix einfügen (nur an den Positionen 1–4, vom Aminoende aus gesehen, ist dies möglich). Wo Prolin auftritt, kommt es zu Abweichungen von der regelmäßigen Struktur. α-Helices sind ziemlich stabil und können als starre Zylinder eine Art Skelett des Proteins bilden. Deshalb findet man sie in der Darstellung von Proteinstrukturen nicht nur als Helix, sondern auch als Zylinder abgebildet. Ein Protein mit überwiegender Helixstruktur ist das Myoglobin, ein dem Hämoglobin verwandtes Muskelprotein.
Geometrie der Helix und Helix-Helix-Wechselwirkungen
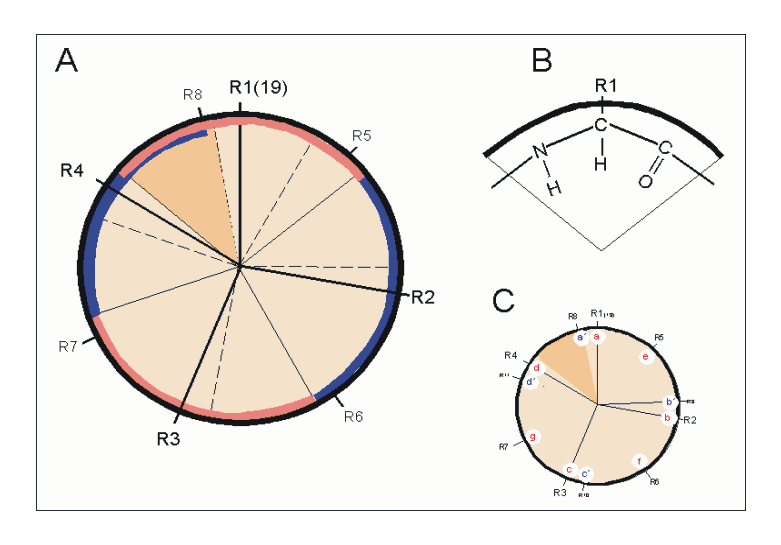
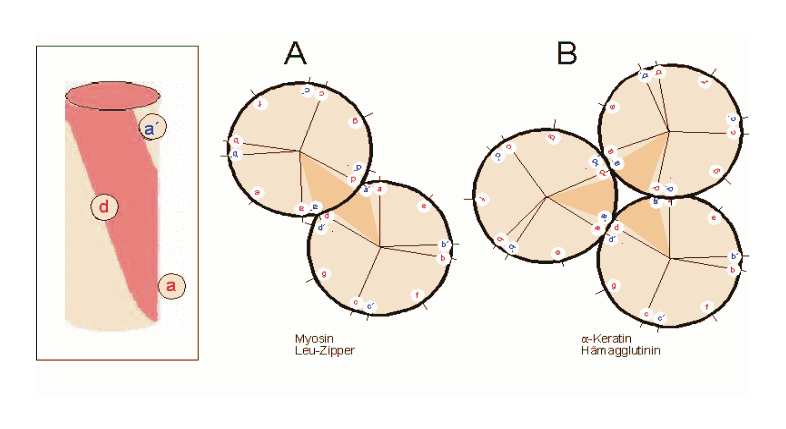
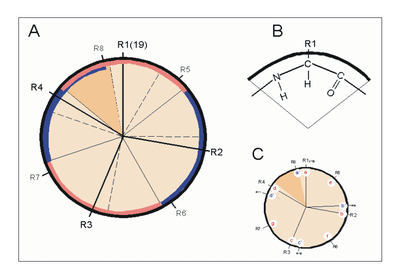 Helixrad (´helical wheel'). Die Endansicht (Projektion) der α-Helix verdeutlicht die Wechselbeziehungen der Aminosäurereste. Wie unter (B) dargestellt, überdeckt jede Aminosäure einen Sektor von 100° [dünne Linien in (A)]. Die nach Abb. 1 Wasserstoff-verbrückten Reste 1 und 5 sind sich – als Teile aufeinanderfolgender Windungen – räumlich nahe. Ähnliches gilt für die Reste 1 und 4 (sog. „n+/-3,4 Kriterium“ gemäß Tab. 2). Während erst der 19. Rest (R19) ekliptisch über R1 zu liegen kommt, gilt dies angenähert bereits für den zwei Windungen entfernten Rest 8; man spricht hier von einem „Pseudorepeat“ (Heptadenrepeat) der Form abcdefga’b’c’d’e’f’g’, wobei a dem Rest 1 und a’ dem Rest 8 entspricht. Sind a und a’ bzw. d und d’ hydrophobe Reste, so entsteht ein linksgewundenes „hydrophobes Band“ um den Zylinder der (rechtsgängigen) α-Helix herum. Hierdurch werden, gemäß Abb. 3, Überstrukturen („Coiled-Coils“) ermöglicht
Helixrad (´helical wheel'). Die Endansicht (Projektion) der α-Helix verdeutlicht die Wechselbeziehungen der Aminosäurereste. Wie unter (B) dargestellt, überdeckt jede Aminosäure einen Sektor von 100° [dünne Linien in (A)]. Die nach Abb. 1 Wasserstoff-verbrückten Reste 1 und 5 sind sich – als Teile aufeinanderfolgender Windungen – räumlich nahe. Ähnliches gilt für die Reste 1 und 4 (sog. „n+/-3,4 Kriterium“ gemäß Tab. 2). Während erst der 19. Rest (R19) ekliptisch über R1 zu liegen kommt, gilt dies angenähert bereits für den zwei Windungen entfernten Rest 8; man spricht hier von einem „Pseudorepeat“ (Heptadenrepeat) der Form abcdefga’b’c’d’e’f’g’, wobei a dem Rest 1 und a’ dem Rest 8 entspricht. Sind a und a’ bzw. d und d’ hydrophobe Reste, so entsteht ein linksgewundenes „hydrophobes Band“ um den Zylinder der (rechtsgängigen) α-Helix herum. Hierdurch werden, gemäß Abb. 3, Überstrukturen („Coiled-Coils“) ermöglicht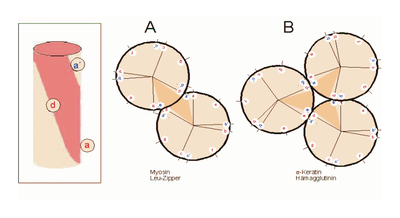 Amphipathische α-Helices können sich zu Überstrukturen, sog. „coiled-coils“ vereinigen. Grundlage sind „hydrophobe Bänder“ (links dargestellt), die immer dann entstehen, wenn die Aminosäuren a, d, a’, d’ in der in Abb. 2 genannten Heptade hydrophob sind. Teil (A) zeigt ein „coiled coil“ aus zwei, Teil (B) ein solches aus drei α-Helices (Projektion). Darüber hinaus wurden „tetrahelix bundle“-Strukturen beschrieben
Amphipathische α-Helices können sich zu Überstrukturen, sog. „coiled-coils“ vereinigen. Grundlage sind „hydrophobe Bänder“ (links dargestellt), die immer dann entstehen, wenn die Aminosäuren a, d, a’, d’ in der in Abb. 2 genannten Heptade hydrophob sind. Teil (A) zeigt ein „coiled coil“ aus zwei, Teil (B) ein solches aus drei α-Helices (Projektion). Darüber hinaus wurden „tetrahelix bundle“-Strukturen beschrieben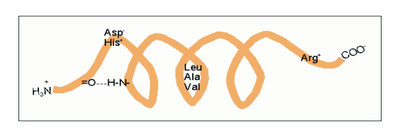 Nukleation und Ausbreitung von α-Helices. Die Bildung einer α-Helix beginnt dort, wo die Reste mehrerer Helixbildner zusammenkommen, darunter insbesondere Leucin, Alanin und Valin. Von diesem Nukleationszentrum ausgehend breitet sich die Struktur aus, bis ein Abbruchsignal erreicht wird. Das stringenteste Abbruchsignal am N-Terminus ist, wie oben angesprochen, ein Prolinrest.
Nukleation und Ausbreitung von α-Helices. Die Bildung einer α-Helix beginnt dort, wo die Reste mehrerer Helixbildner zusammenkommen, darunter insbesondere Leucin, Alanin und Valin. Von diesem Nukleationszentrum ausgehend breitet sich die Struktur aus, bis ein Abbruchsignal erreicht wird. Das stringenteste Abbruchsignal am N-Terminus ist, wie oben angesprochen, ein Prolinrest.α-Helices sind die Grundlage typischer Faserproteine (α-Keratin, der Grundsubstanz der Haare, Myosin, einer Komponente der Muskelfasern usw.) aber auch, wie am Beispiel des Myoglobins eingeführt, strukturgebende Komponenten von löslichen, globulären Proteinen. Einzelne Helices können diese Aufgabe im Allgemeinen nicht übernehmen, wohl aber geordnete Aggregate aus zwei, drei, vier oder mehr Individualhelices. Abb. 3 veranschaulicht, wie sich zwei bzw. drei α-Helices aufgrund hydrophober Wechselwirkungen zu einer „Superhelix“ zusammenlagern können. Dies setzt amphipathische Helices voraus, das sind Helices deren eine Seite hydrophil (dem Wasser zugewandt) und deren andere Seite hydrophob und damit zu Wechselwirkungen befähigt ist. Die in Abb. 1 gezeigte Geometrie bewirkt dabei, dass das „hydrophobe Band“ nicht parallel zur Helixachse verläuft, sondern die Helix in Form einer gedehnten, linksgängigen Spirale umgibt. Wenn sich die hydrophoben Bänder von zwei oder mehr Helices nähern, entsteht die als „Coiled-Coil“ bezeichnete Superhelix.
Helix-Vorhersage
Erste Bemühungen zur Vorhersage von Protein-Sekundärstrukturen gehen auf die sechziger Jahre des vergangenen Jahrhunderts zurück und konnten mit dem Aufkommen der modernen Röntgenstrukturanalyse kontinuierlich verfeinert werden. Ein überaus hilfreicher, rationaler Ansatz zur Vorhersage der α-Helix verbindet sich mit dem Namen Marianne Schiffer und schließt an die obigen Überlegungen an. So illustriert Abb. 1 das n+/-3,4 Kriterium, wonach ein Rest n mit Resten paaren kann, die drei bzw. vier Positionen entfernt sind. Sind so z. B. die Reste 1, 4 und 5 hydrophob, so können sie wechselwirken und damit eine Helixstruktur stabilisieren. Gleiches gilt für die Reste 6, 3 und 2 usw. Dieses Vorhersageschema zeigte zunächst für Insulin und Myoglobin seinen Wert.
Mit der Veröffentlichung weiterer Röntgenstrukturanalysen wich der „helical-wheel“-Ansatz zunehmend statistischen Verfahren. Ein früher Ansatz dieser Art geht auf Chou und Fasman (1974, 1978) zurück. Nachfolgend wird eine Tabelle wiedergegeben, die die Helixpotentiale von Aminosäureresten wiedergibt. Dabei spricht man von „Helixbildnern“, wenn das Potential (Pα) deutlich über 1 liegt und von „Helixbrechern“, wenn es deutlich kleiner ist.
Aminosäure Pα Pαi Glu 1,53 1,45 Ala 1,45 1,59 Leu 1,34 1,91 His 1,24 0,87 Met 1,20 1,25 Gln 1,17 0,98 Trp 1,14 1,33 Val 1,14 1,42 Phe 1,12 1,12 Lys 1,07 1,13 Ile 1,00 1,22 Asp 0,98 0,53 Thr 0,82 0,75 Ser 0,79 0,70 Arg 0,79 0,67 Cys 0,77 0,33 Asn 0,73 0,53 Tyr 0,61 0,58 Pro 0,59 0 Gly 0,53 0,53 Helixpotentiale (Pα und Pαi) der Aminosäurereste. Pα entspricht der Wahrscheinlichkeit, mit der ein Rest generell in der Helix vertreten ist, Pαi ist die Wahrscheinlichkeit, mit der er im Inneren der Helix vorkommt. Bemerkenswert ist die Tatsache, dass Prolin einen positiven Wert für Pα aufweist, im Zentrum einer Helix jedoch nicht existieren kann (Pαi = 0). Wie in Abb. 1 dargestellt fungieren die Reste 1–4 einer Helix als Akzeptoren einer H-Brücke, ab Rest 5 jedoch gleichzeitig als Wasserstoffdonoren. Diese Funktion scheidet bei Pro aufgrund einer fehlenden NH2-Gruppe aus. Zusammenfassung der Helix-Parameter
A – Relation der Reste zueinander n + 4 Reste 1 und 5 H-Brücke -C=O···HN- n +/− 3, 4 Reste 1 und 4 oder 5 gleiche Seite „hydrophober Bogen“, α-Potenzial n + 18 Reste 1 und 19 ekliptisch 5 × 3,6 = 18; „repeat unit“ n + 7 Reste 1 und 8 „fast ekliptisch“ 2 × 3,6; „Heptadenrepeat“ B – Physikalische Parameter n = 3,6 Reste pro Windung d = 1,5 A axiale Verschiebung je Rest p = 5,4 Å = n x d “Pitch” (Abstand zwischen den Windungen) a = 100° = 360° / n Winkel (Sektor) je Aminosäure Literatur
- Schiffer, M. & Edmundson, A.B. (1967): Use of helical wheels to represent the structrures of proteins and to identify segments with helical potential. Biophys. J. 7, 121–135.
- Chou, P.Y. & Fasman, G.D. (1978): Empirical predictions of protein conformation. Ann. Rev. Biochem. 47, 251–276.
- Cohen, C. and Parry, D.A.D. (1986): α-Helical coiled-coils – a widespread motif in proteins. Trends in Biochem. Sci. 11, 245–248.
- Kamtekar, S., Schiffer, J. M., Xiong, H., Babik, J. M. & Hechtr, M. H. (1993): Protein design by binary patterning of polar and nonpolar amino acids. Science 262, 1680–1685.
Siehe auch
Wikimedia Foundation.