- Lysosomale α-Glucosidase
-
Lysosomale α-Glucosidase —
Masse/Länge Primärstruktur 883 Aminosäuren Bezeichner Gen-Name GAA Externe IDs OMIM: 606800 UniProt: P10253 Enzymklassifikation EC, Kategorie 3.2.1.20 Glykosidase Reaktionsart Hydrolyse Substrat endständige 1,4-verbundene α-D-Glucosereste Produkte α-D-Glucose Vorkommen Homologie-Familie saure Maltase Übergeordnetes Taxon Euteleostomi Die lysosomale α-Glucosidase (auch saure Maltase, Gen: GAA) ist dasjenige Enzym, das in Lysosomen langkettige Polysaccharide zu Glucose abbaut. Es ist kein Teil des Glykogenabbaus in der Leber und auch kein Teil der Verdauung von Polysacchariden im Darm, sondern hilft beim Abbau von Fremdstoffen in den Lysosomen. Die saure Maltase kommt in Wirbeltieren vor. Beim Menschen ist sie in allen Gewebetypen lokalisiert. Mutationen am GAA-Gen können zur Glykogenspeicherkrankheit Typ II (Morbus Pompe) führen.[1]
Ein potenter Hemmstoff der sauren Maltase ist das natürlich vorkommende Salacinol.[2]
Katalysierte Reaktion
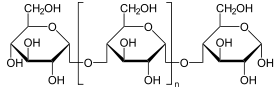 (n=m) + H2O
(n=m) + H2O 
(n=m-1) + GlucoseEndständige Glucose wird von Polysacchariden abgespalten.
Einzelnachweise
- ↑ UniProt P10253
- ↑ Minami Y, Kuriyama C, Ikeda K, et al: Effect of five-membered sugar mimics on mammalian glycogen-degrading enzymes and various glucosidases. In: Bioorg. Med. Chem.. 16, Nr. 6, März 2008, S. 2734–40. doi:10.1016/j.bmc.2008.01.032. PMID 18258441.
Weblinks
 Wikibooks: Biochemie und Pathobiochemie: Glycogenolyse und Stärkeabbau – Lern- und LehrmaterialienKategorien:
Wikibooks: Biochemie und Pathobiochemie: Glycogenolyse und Stärkeabbau – Lern- und LehrmaterialienKategorien:- Glykosidase
- Krankheitsassoziiertes Protein

Wikimedia Foundation.