- Apert-Syndrom
-
Klassifikation nach ICD-10 Q87.0 Angeborene Fehlbildungssyndrome mit vorwiegender Beteiligung des Gesichtes
- Akrozephalosyndaktylie-Syndrome (Apert)ICD-10 online (WHO-Version 2011) Das Apert-Syndrom, auch Akrozephalosyndaktylie genannt, ist eine genetisch bedingte Besonderheit auf der Grundlage einer Mutation des FGFR2-Gens auf dem Chromosom 10, die zu vielfältigen körperlichen Fehlbildungen führt. Benannt wurde das Syndrom 1896 nach dem französischen Kinderarzt Eugene Apert. Es gehört zur Gruppe der kraniofazialen Fehlbildungen, zu der auch das Carpenter-Syndrom, das Crouzon-Syndrom, das Pfeiffer-Syndrom und das Saethre-Chotzen-Syndrom gehören.
Inhaltsverzeichnis
Häufigkeit
In Deutschland leben etwa 400 Personen mit dem Syndrom. Die Inzidenz beträgt etwa 1:1,000,000, für alle fünf Syndrome zusammen bei 1:90.000.
Symptome und Merkmale
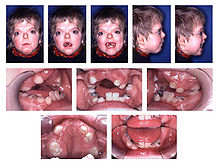 Erscheinungsbild und Fehlbildungen beim Apert-Syndrom
Erscheinungsbild und Fehlbildungen beim Apert-Syndrom
- Kopfbereich:
- Verwachsen von Schädelknochen mit der Gefahr eines Drucks auf das Gehirn und der Bildung eines Hydrocephalus
- Fehlbildung des Oberkiefers
- offene Gaumenspalte
- eingeschränktes Hörvermögen
- Sehbehinderung
- behinderte Atmung
- Extremitäten:
- zusammengewachsene Finger und Zehen (tritt stets symmetrisch auf)
- Versteifte oder fehlende Mittelgelenke
- Knochenbau:
- Eingeschränkte Bewegung in vielen Gelenken
- Verkrümmung der Wirbelsäule (Skoliose)
Das Syndrom hat mitunter schwere Auswirkungen auf die soziale und emotionale Entwicklung eines Kindes ("anders sein"). Sofortige bzw. baldmöglichste Behandlung durch einen Spezialisten sind für die Linderung extrem wichtig. So z.B.
- Schädeloperationen, welche dem Gehirn genügend Platz verschaffen
- Eingriffe ins Mittelohr (fehlende Knochen)
- Trennungsoperationen an Händen und Füßen.
In der Pränataldiagnostik fallen typische Merkmale oft bereits in frühen Stadien der Schwangerschaft auf, häufig im Rahmen des Feinultraschalls. Eine Diagnose ist vorgeburtlich durch die Untersuchung des Fruchtwassers möglich. Nachgeburtlich sind die meisten körperlichen Merkmale offensichtlich, eine molekulargenetische Analyse kann den Befund bestätigen.
Erbgang
Der Erbgang ist autosomal-dominant mit kompletter Penetranz und hoher Varianz (die Ausprägung gestaltet sich in Abhängigkeit von der Mutation und dem Individuum sehr variabel)
- Fibroblast growth factor receptor (FGFR2)-Gen[1]
- verschiedene Mutationen aktivieren den Rezeptor, das führt zum verstärkten Signal, und damit zu Veränderungen vor allem in der Knochen- und Knorpelgewebe Differenzierung
- 80 % Neumutationen, väterlicher Alterseffekt
Die Hypochondroplasie (leichtere Manifestationsform) und die Thanatophore Dysplasie (schwerere und letale Manifestationsform) zeigen Mutationen im gleichen FGFR3-Gen.
Einzelnachweise
- ↑ APERT SYNDROME. auf: omim.org
Weblinks

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Fehlbildung
- Erbkrankheit
- Krankheitsbild in der Kinderheilkunde
- Kopfbereich:

Wikimedia Foundation.