- Reissert-Reaktion
-
Die Reissert-Reaktion ist eine Namensreaktion der organischen Chemie, welche die Funktionalisierung sechsgliedriger Stickstoffaromaten, generell Pyridin oder benzannelierte Heterozyklen wie Chinolin und Isochinolin, ermöglicht.[1],[2]
Inhaltsverzeichnis
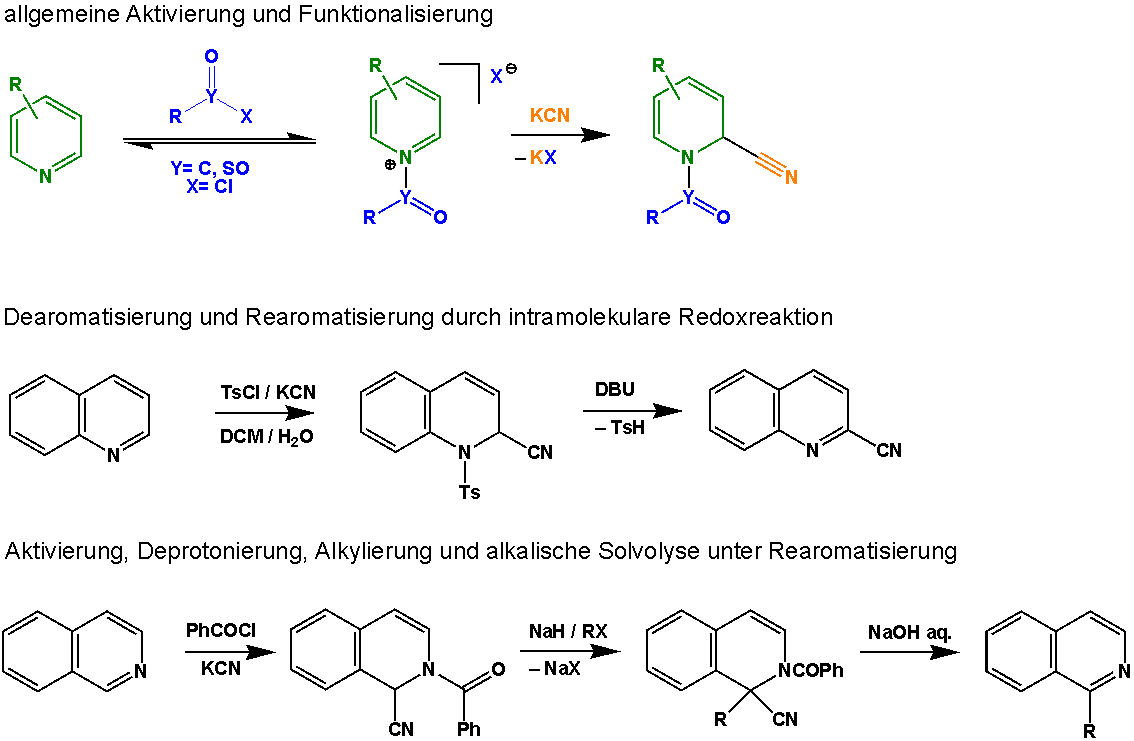
Reaktionsmechanismus
Die initiale Aktivierung des Aromaten wird zumeist durch Umsetzung der aromatischen Base mit einem Säurechlorid erreicht (sowohl Carbonsäurechloride als auch Sulfonsäurechloride). Die Acylierung des aromatischen Stickstoffatoms bewirkt eine Steigerung der Elektrophilie des Systems, sodass das gebildete Ammoniumion (speziell Pyridinium, Chinolinium oder Isochinolinium) durch das Cyanid-Nucleophil attackiert werden kann. Dieser nucleophile Angriff führt zur Dearomatisierung des Systems unter Bildung einer Dienstruktur. Wird als Aktivierungsreagenz ein Sulfonsäurechlorid benutzt, kann der aromatische Charakter des Systems unter Einwirkung einer nichtnucleophilen Base (bspw. DBU) durch intramolekulare Redoxprozesse wiederhergestellt werden. Dabei wird die Sulfonylgruppe (im dargestellten Beispiel Ts) zur entsprechenden Sulfinsäure reduziert (TsH entspricht dabei TolSO2H, para-Toluolsulfinsäure) und als Produkt ein mit einer Cyanogruppe funktionalisiertes Derivat erhalten. Bei Einsatz von Carbonsäurechloriden kann das cyanierte N-Acyl-Derivat mit starken Basen deprotoniert (bspw. NaH) und durch Alkylierung und nachgestellte alkalische Hydrolyse in viele weitere Derivate überführt werden. Dabei macht man sich die Acidifizierung der Methinfunktion durch die Nitrilgruppe zunutze.
Überblick
Anwendung
Die Reissert-Reaktion dient bspw. zur Synthese von Isochinolinderivaten wie dem Praziquantel.
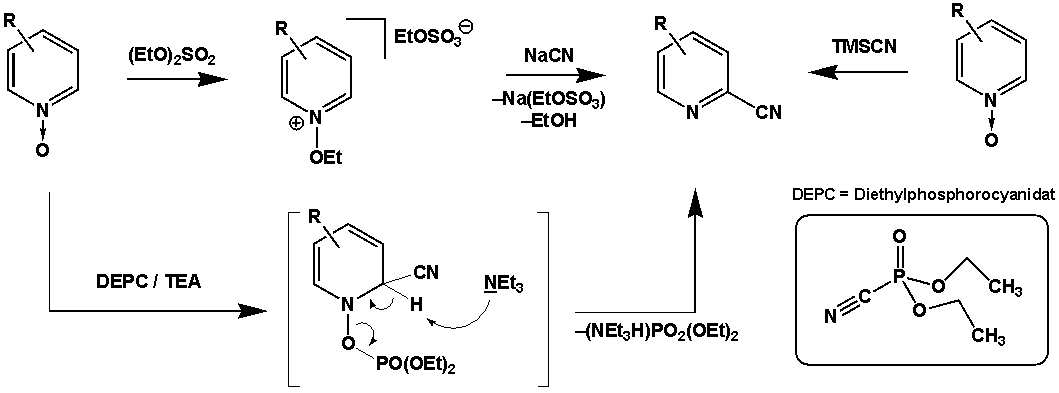
Reissert-Henze-Reaktion
Ebenso wie die Reissert-Reaktion selbst, ermöglicht die Reissert-Henze-Reaktion (auch als Reissert-Kaufmann Reaktion[3] bekannt) die Synthese von heterozyklischen Cyanoaromaten. Im Gegensatz zur Reissert-Reaktion dienen jedoch N-Oxide als Ausgangsmaterial. An Pyridin-N-Oxiden ist die Reissert-Reaktion selbst zumeist nicht erfolgreich, Ausnahmen existieren jedoch (bspw. 4-Chlorpyridin-N-Oxid).[4] Die Rearomatisierung wird ähnlich wie bei der Boekelheide-Umlagerung durch den Bruch der NO-Bindung erreicht. Hierzu muss die N-Oxid Funktion zunächst aktiviert werden. Im klassischen Protokoll dient dazu die Alkylierung mit Dialkylsulfaten (Vgl. Dimethylsulfat), es existieren aber auch Modifikationen, die die Aktivierung durch Silylierung (Vgl. Trimethylsilylcyanid) oder Phosphorylierung (Vgl. Diethylphosphorocyanidat) ermöglichen.[5]
Einzelnachweise
- ↑ Thomas L. Gilchrist: Heterocyclic Chemistry. 3. Auflage. 1998, ISBN 0-582-27843-0, S. 169.
- ↑ J.A. Joule, K. Mills: Heterocyclic Chemistry. 4. Auflage. 2004, ISBN 0-632-05453-0, S. 131 f.
- ↑ R. A. Abramovitch, Erwin Klingsberg: Pyridine and its Derivatives. Supplement, Teil 2. (Part 2, Volume 14), ISBN 0-471-37914-X, S. 117. (The Chemistry of Heterocyclic Compounds. v. 14)
- ↑ R. A. Abramovitch, Erwin Klingsberg: Pyridine and its Derivatives. Supplement, Teil 2. (Part 2, Volume 14), ISBN 0-471-37914-X, S. 114. (The Chemistry of Heterocyclic Compounds. v. 14)
- ↑ Jie Jack Li: Name Reactions in Heterocyclic Chemistry. 1. Auflage. 2005, ISBN 0-471-30215-5, S. 345 ff.
Wikimedia Foundation.