- Fechtner-Syndrom
-
Klassifikation nach ICD-10 D69.1 Qualitative Thrombozytendefekte ICD-10 online (WHO-Version 2011) Das Fechtner-Syndrom ist eine sehr seltene autosomal-dominante Erbkrankheit. Sie wird durch eine Mutation des MYH9-Gens verursacht. Das Fechtner-Syndrom gehört zusammen mit dem Sebastian-Syndrom, dem Epstein-Syndrom und der May-Hegglin-Anomalie zur Gruppe der MYH9-assoziierten Erkrankungen.
Inhaltsverzeichnis
Ursache und Genetik
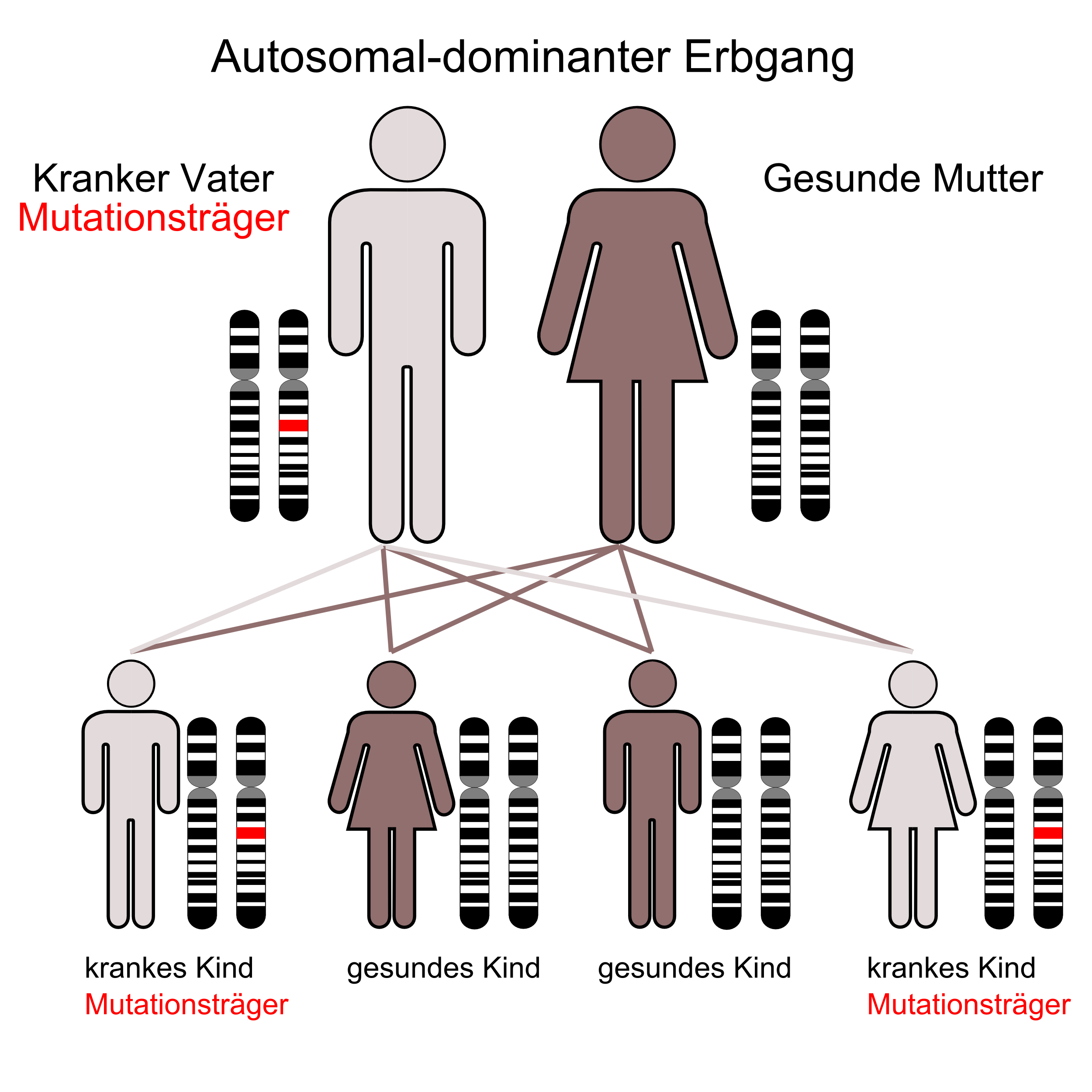
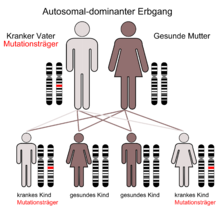 Der autosomal-dominante Erbgang
Der autosomal-dominante Erbgang
Die Ursache des Fechtner-Syndroms sind Punktmutationen des MYH9-Gens, das sich beim Menschen auf Chromosom 22 Genlocus q11.2 befindet.[1][2][3]
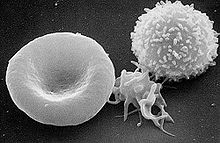
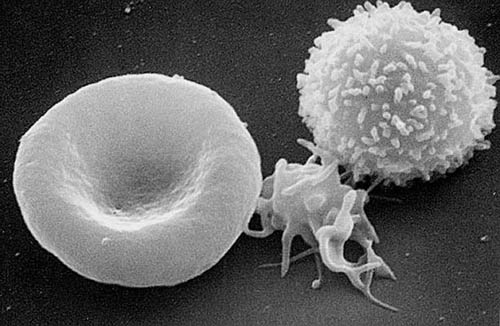
Das Gen kodiert für die schwere Kette eines Nicht-Muskel-Myosins Typ IIA (NMMHC-IIA). Dieses Protein wird in einigen Blutzellen, unter anderem in Monozyten und Thrombozyten, in der Hörschnecke (Cochlea) und in den Nieren exprimiert. Die Mutation bewirkt eine Störung der Dimerisierung des NMMHC-IIA-Proteins. Die Folge davon ist eine Instabilität und eine Präzipitation zusammen mit normalen MYHIIA-Ketten im Zytoplasma der Leukozyten. Diese Präzipitate bilden die Zytoplasmaeinschlüsse. Die Störung der Dimerisierung bewirkt zudem eine fehlerhafte Organisation des Zytoskeletts in den Megakaryozyten, den Vorläuferzellen der Thrombozyten. Dies ist die Ursache für die Makrothrombozytopenie, die sich durch einen Mangel an Thrombozyten (eine sogenannte Thrombozytopenie) und übergroßen Thrombozyten mit Leukozyteneinschlüssen manifestiert. Die Größe der Thrombozyten kann dabei die von Erythrozyten sogar übertreffen.[4]
Epidemiologie und Prävalenz
Aufgrund der Seltenheit des Fechtner-Syndroms sind keine gesicherten Daten bezüglich der Epidemiologie und der Prävalenz verfügbar.[4] Die Prävalenz liegt aber deutlich unter 1:100.000.
Symptome und Diagnose
Entsprechend den Stellen der Genexpression von NMMHC-IIA manifestierten sich auch die Symptome des Fechtner-Syndroms. So zeigen die vom Fechtner-Syndrom betroffenen Patienten eine Makrothrombozytopenie mit Leukozyteneinschlusskörpern. Die Einschlusskörper in den Thrombozyten sind relativ klein und mit einer Routinefärbung meist schwer zu erkennen. Immunfluoreszenz-Methoden sind zum Nachweis besser geeignet.[4]
Etwa die Hälfte der Patienten entwickelt einen progredienten Hörverlust (49%).[5]
Bei einigen Patienten kann sich zudem eine Glomerulonephritis (eine Entzündung der Nierenfilterchen), sowie ein Grauer Star einstellen. Beide Erkrankungen stellen sich aber nicht zwangsläufig ein, da die Dimerisierungs-Störung von NMMHC-IIA durch eine andere Myosinform (Typ IIB) ausgeglichen werden kann.[4]
Die meisten Patienten leiden – im Gegensatz zum Sebastian-Syndrom – nicht unter verstärkten Blutungen.
Therapie
Die Therapie erfolgt im Wesentlichen symptomatisch. Vor Operationen ist unter Umständen eine Thrombozytentransfusion notwendig.
Entdeckung
Das Fechtner-Syndrom wurde erstmals 1985 von L. C. Peterson et al. beschrieben.[6][7]
Einzelnachweise
- ↑ UCSC Genome Browser on Human Mar. 2006 Assembly: chr22:35,007,273-35,113,958
- ↑ genome.ucsc.edu: Human Gene MYH9 (uc003apg.1) Description and Page Index
- ↑ A. Toren u. a.: Genetic linkage of autosomal-dominant Alport syndrome with leukocyte inclusions and macrothrombocytopenia (Fechtner syndrome) to chromosome 22q11-13. In: Am. J. Hum. Genet. 65/1999, S.1711–7. PMID 10577925
- ↑ a b c d C. Trichet: Fechtner-Syndrom Oktober 2006
- ↑ W. Delb u. a.: Das Fechtner-Syndrom: Eine seltene Differentialdiagnose des Alport-Syndroms. In: HNO, 48/2000, S.616–20.
- ↑ L. C. Peterson u. a.: Fechtner syndrome: a variant of Alport's syndrome with leukocyte inclusions and macrothrombocytopenia. In: Blood 65/1985, S.397–406. PMID 2981587
- ↑ R. Gershoni-Baruch u. a.: Fechtner syndrome: clinical and genetic aspects. In: Am. J. Med. Genet. 31/1988, S.357–67. PMID 3232700
Literatur
- S. Deutsch u. a.: Asp1424Asn MYH9 mutation results in an unstable protein responsible for the phenotypes in May-Hegglin anomaly/Fechtner syndrome. In: Blood 102/2003, S.529–34. PMID 12649151
- J. G. Drachman: Inherited thrombocytopenia: when a low platelet count does not mean ITP. In: Blood, 103/2004, S.390–398.
- R. Gershoni-Baruch u. a.: Fechtner syndrome: clinical and genetic aspects. In: Am. J. Med. Genet. 31/1988, S.357–67. PMID 3232700
- T. F. Leung u. a.: A Chinese adolescent girl with Fechtner-like syndrome. In: Acta Paediatr 87/1998, S.705–7.
- P. Mhawech, S. Abdus: Inherited Giant Platelet Disorders: Classification and Literature Review. In: American Journal of Clinical Pathology 2000, S.176–90.
- N. Pujol-Moix u. a.: Ultrastructural analysis of granulocyte inclusions in genetically confirmed MYH9-related disorders. In: Haematologica, 89/2004, S.330–7. PMID 15020273
- M. Saito u. a.: Hematological abnormalities in a patient with a 22q11.2 deletion. In: Brain Dev 26/2004, S.342–4. PMID 15165677
- M. Seri u. a.: Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner Syndrome Consortium. In: Nature Genetics 26/2000, S.103–5. PMID 10973259
- M. Seri u. a.: MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. In: Medicine (Baltimore) 82/2003, S.203–15. PMID 12792306
Weblinks
- Fechtner-Syndrom bei Online Mendelian Inheritance in Man
- genome.ucsc.edu: Human Gene MYH9 (uc003apg.1) Description and Page Index (engl.)

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in Hämatologie und Onkologie
- Erbkrankheit
Wikimedia Foundation.