- May-Hegglin-Anomalie
-
Klassifikation nach ICD-10 D72.0[1] Genetisch bedingte Leukozytenanomalien ICD-10 online (WHO-Version 2011) Die May-Hegglin-Anomalie (MHA) ist eine sehr seltene autosomal-dominante Erbkrankheit, bei der die Blutplättchen verändert sind. Sie wird durch eine Mutation des MYH9-Gens verursacht. Die May-Heggelin-Anomalie gehört zusammen mit dem Sebastian-Syndrom, dem Fechtner-Syndrom und dem Epstein-Syndrom zur Gruppe der MYH9-assoziierten Erkrankungen. Die MHA ist von diesen ausgesprochen seltenen Erkrankungen die häufigste Form.[2]
Inhaltsverzeichnis
Ursache und Genetik
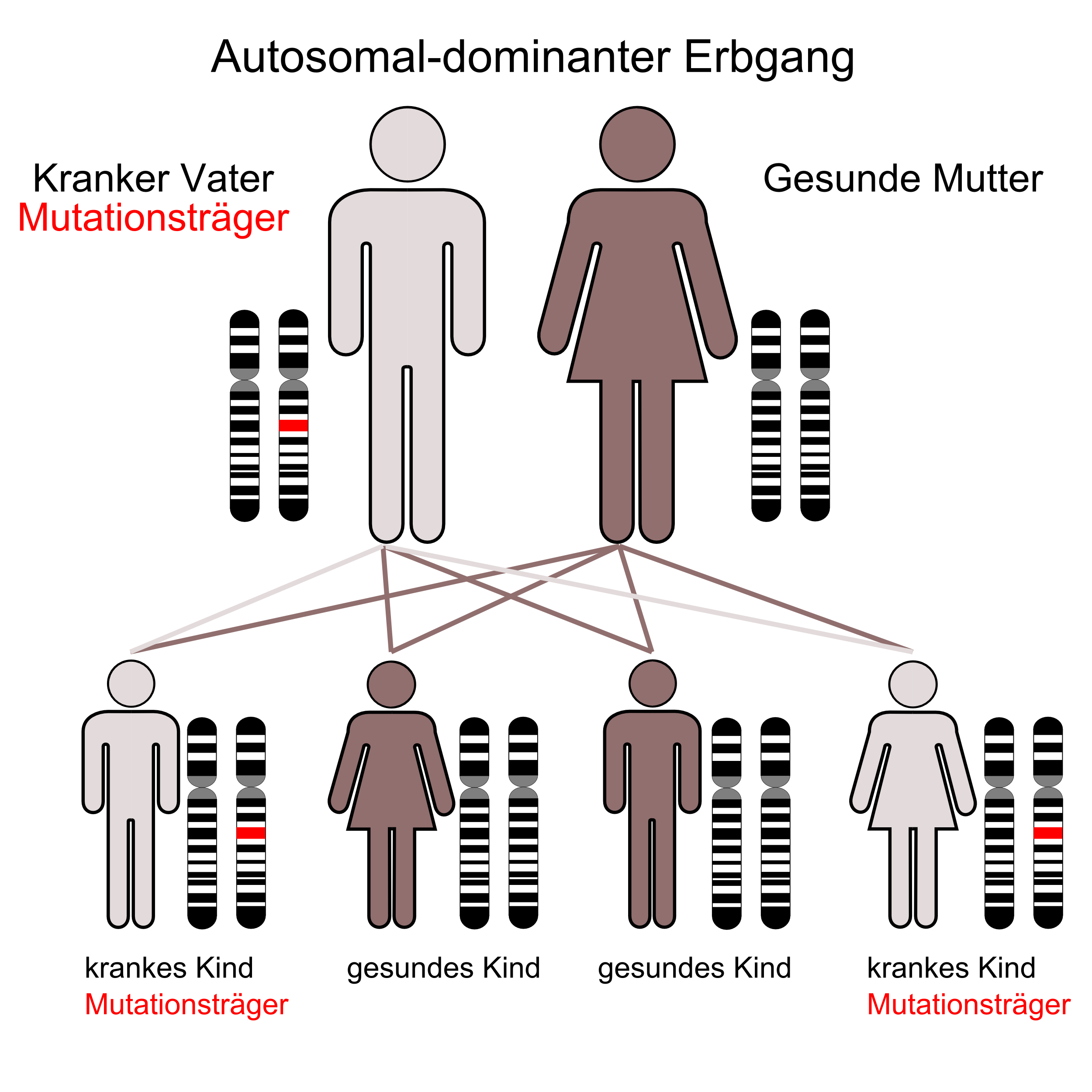
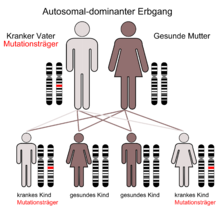 Der autosomal-dominante Erbgang
Der autosomal-dominante Erbgang
Die Ursache der MHA ist eine Punktmutation des MYH9-Gens, das sich beim Menschen auf Chromosom 22 Genlocus q11.2 befindet.[3][4][5]
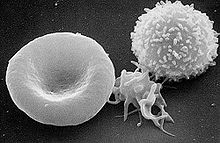
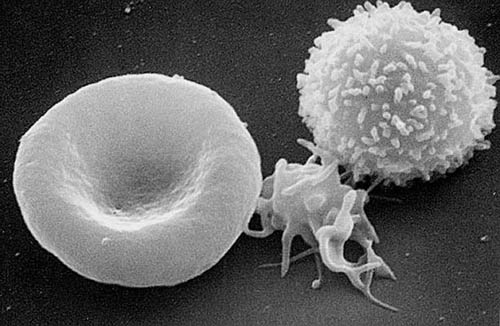
Das Gen kodiert für die schwere Kette eines Nicht-Muskel-Myosins Typ IIA (NMMHC-IIA). Dieses Protein wird in einigen Blutzellen, unter anderem in Monozyten und Thrombozyten, in der Hörschnecke (Cochlea) und in den Nieren exprimiert. Die Mutation bewirkt offensichtlich eine Konformationsänderung im Kopf des NMMHC-IIA-Proteins. Die Folge davon ist eine Störung der Aggregation des Proteins zu Döhle-Körperchen, was eine fehlerhafte Organisation des Zytoskeletts in den Megakaryozyten, den Vorläuferzellen der Thrombozyten bewirkt. Dies ist die Ursache für die Makrothrombozytopenie, die sich durch einen Mangel an Thrombozyten (eine sogenannte Thrombozytopenie) und übergroßen Thrombozyten mit Leukozyteneinschlüssen manifestiert. Die Größe der Thrombozyten kann dabei die von Erythrozyten sogar übertreffen.[6]
Symptome und Diagnose
MHA-Patienten weisen einen Mangel an Thrombozyten (Thrombozytopenie), sowie zusätzlich eine Funktionsstörung der Thrombozyten (Thrombozytopathie) auf.[7] Die vorhandenen Thrombozyten sind erheblich größer als bei nicht betroffenen Menschen (Makrothrombozytopenie).
Im Gegensatz zum Epstein- und Fechtner-Syndrom entwickeln die Patienten keinen Hörverlust und keine Glomerulonephritis.[8]
Therapie
Die Therapie erfolgt im Wesentlichen symptomatisch. Vor Operationen ist unter Umständen eine Thrombozytentransfusion notwendig.[9]
Epidemiologie und Prävalenz
Aufgrund der Seltenheit der May-Hegglin-Anomalie sind keine gesicherten Daten bezüglich der Epidemiologie und der Prävalenz verfügbar. Die Prävalenz wird auf 1:500.000 geschätzt.[9]
Prognose
Die meisten Patienten haben eine normale Lebenserwartung.[9]
Entdeckung
Die May-Heggelin-Anomalie wurde erstmals 1909 von dem Münchner Internist Richard May (1863–1937) bei einer 24 Jahre alten Patientin beschrieben. Er fand in ihrem peripheren Blut Einschlüsse in den Leukozyten.[10] 1945 berichtete der Schweizer Internist Robert Hegglin (1907–1969) von Riesenthrombozyten mit Thrombozytopenie und Einschlüssen in Leukozyten bei zwei Generationen einer Familie.[11]
Einzelnachweise
- ↑ ICD-Code: D70-D77 Sonstige Krankheiten des Blutes und der blutbildenden Organe.
- ↑ M. Picu u. a.: An 8-Year-Old Girl With Thrombocytopenia In: Archives of Pathology and Laboratory Medicine 129/2005, S.214–7.
- ↑ UCSC Genome Browser on Human Mar. 2006 Assembly: chr22:35,007,273-35,113,958
- ↑ genome.ucsc.edu: Human Gene MYH9 (uc003apg.1) Description and Page Index
- ↑ J. A. Martignetti u. a.: The gene for May-Hegglin anomaly localizes to a <1-Mb region on chromosome 22q12.3-13.1. In: Am J Hum Genet 66/2000, S.1449–54. PMID 10739770
- ↑ C. Trichet: Thrombozytopenie May-Hegglin Oktober 2006
- ↑ R. E. Scharf: Angeborene und erworbene Thrombozytopenien. In: Hämostaseologie 23/2003, S.159–69.
- ↑ K. E. Heath u. a.: Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. In: Am J Hum Genet 69/2001, S. 1033–45. PMID 11590545
- ↑ a b c National Organization for Rare Disorders: NORD Guide to Rare Disorders. Lippincott Williams & Wilkins, 2003, S. 403. ISBN 0-781-73063-5
- ↑ R. May: Leukozyteneinschlüsse. In: Dtsch. Arch. Klin. Med. 96/1909, S. 1–6.
- ↑ R. Hegglin: Gleichzeitige konstitutionelle Veranderungen an Neutrophilen und Thrombocyten. In: Helv. Med. Acta 12/1945, S. 439–40.
Literatur
- P. Beris u. a.: Post-infection transitory partial correction of thrombocytopenia in may-hegglin anomaly. In: American Journal of Hematology 46/1994, S. 60. PMID 7605407
- J. R. Cabrera u. a.: Defective neutrophil mobility in the May-Hegglin anomaly. In: Br J Haematol 47/1981, S. 337-43. PMID 7459275
- J. G. Drachman: Inherited thrombocytopenia: when a low platelet count does not mean ITP. In: Blood, 103/2004, S. 390–8. PMID 10891439
- E. Jantunen: Inherited giant platelet disorders. In: Eur J Haematol 53/1994, S. 191-6. PMID 7957801
- P. Mhawech, S. Abdus: Inherited Giant Platelet Disorders: Classification and Literature Review. In: American Journal of Clinical Pathology 2000, S. 176–90. PMID 10664620
- P. Noris u. a.: Thrombocytopenia, giant platelets, and leukocyte inclusion bodies (May-Hegglin anomaly): clinical and laboratory findings. In: Am J Med 104/1998, S. 355–60. PMID 9576409
- N. Pujol-Moix u. a.: Ultrastructural analysis of granulocyte inclusions in genetically confirmed MYH9-related disorders. In: Haematologica, 89/2004, S. 330–7. PMID 15020273
- M. Seri u. a.: MYH9-related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. In: Medicine (Baltimore) 82/2003, S. 203–15. PMID 12792306
Weblinks
- May-Hegglin-Anomalie bei Online Mendelian Inheritance in Man
- genome.ucsc.edu: Human Gene MYH9 (uc003apg.1) Description and Page Index
- Universität Lyon: May-Hegglin-Anomalie

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.