- Neuronale Ceroid-Lipofuszinose
-
Klassifikation nach ICD-10 E75.4 Neuronale Zeroidlipofuszinose ICD-10 online (WHO-Version 2011) Die neuronalen Ceroid-Lipofuszinosen (NCL oder CLN), auch als VSS oder veraltet als Amaurotische Idiotie bezeichnet, sind eine Gruppe seltener, autosomal-rezessiv vererbter und bislang noch unheilbarer Stoffwechselkrankheiten, die in unterschiedlichen Formen und Altersstufen auftreten können. Die Krankheit tritt meist im Alter von 1 bis 8 Jahren mit einer maximalen Häufigkeit von 1:12.500 Lebendgeborenen auf. Mittlerweile sind neun NCL-Typen (CLN1-CLN9) bekannt, wobei die Typen CLN1 und CLN4 auch erst im Erwachsenenalter auftreten können. Die neuronalen Ceroid-Lipofuszinosen gehören zu den lysosomalen Speicherkrankheiten.
Die Bezeichnung Neuronale Ceroid-Lipofuszinosen (NCL) leitet sich ab von:
- Neuronen, den Nervenzellen
- Ceroid, einem wachsartigen Stoff
- Lipofuszin, einem fettigen, bräunlichen Stoff
Inhaltsverzeichnis
Geschichte
Die Krankheit wurde erstmals 1826 von dem norwegischen Landarzt Stengel anhand von vier Geschwistern beschrieben und bis 1903 Stengelsche Krankheit genannt. Später war die Bezeichnung Batten-Krankheit (heute noch im englischen Sprachraum als batten disease geläufig) und Vogt-Spielmeyer-Stock-Krankheit (VSS, heute noch in Deutschland verwendet) üblich. Dass es sich um eine Stoffwechselerkrankung handelt, wurde 1939 nachgewiesen. Für die Fette und Proteine, die sich als Abfallstoffe in den Zellen anlagern, wurde 1963 die Bezeichnung Ceroid Lipofuscin Pigment geprägt. Im Jahr 1995 fand man heraus, dass ein Gendefekt - nämlich der Verlust eines Chromosomenabschnittes - ursächlich für die Erkrankung ist. Dass den Betroffenen ein lysosomales Enzym fehlt, welches für den Abbau von Abfallstoffen in den Zellen benötigt wird, ist seit 1998 bekannt.
Krankheitsbild
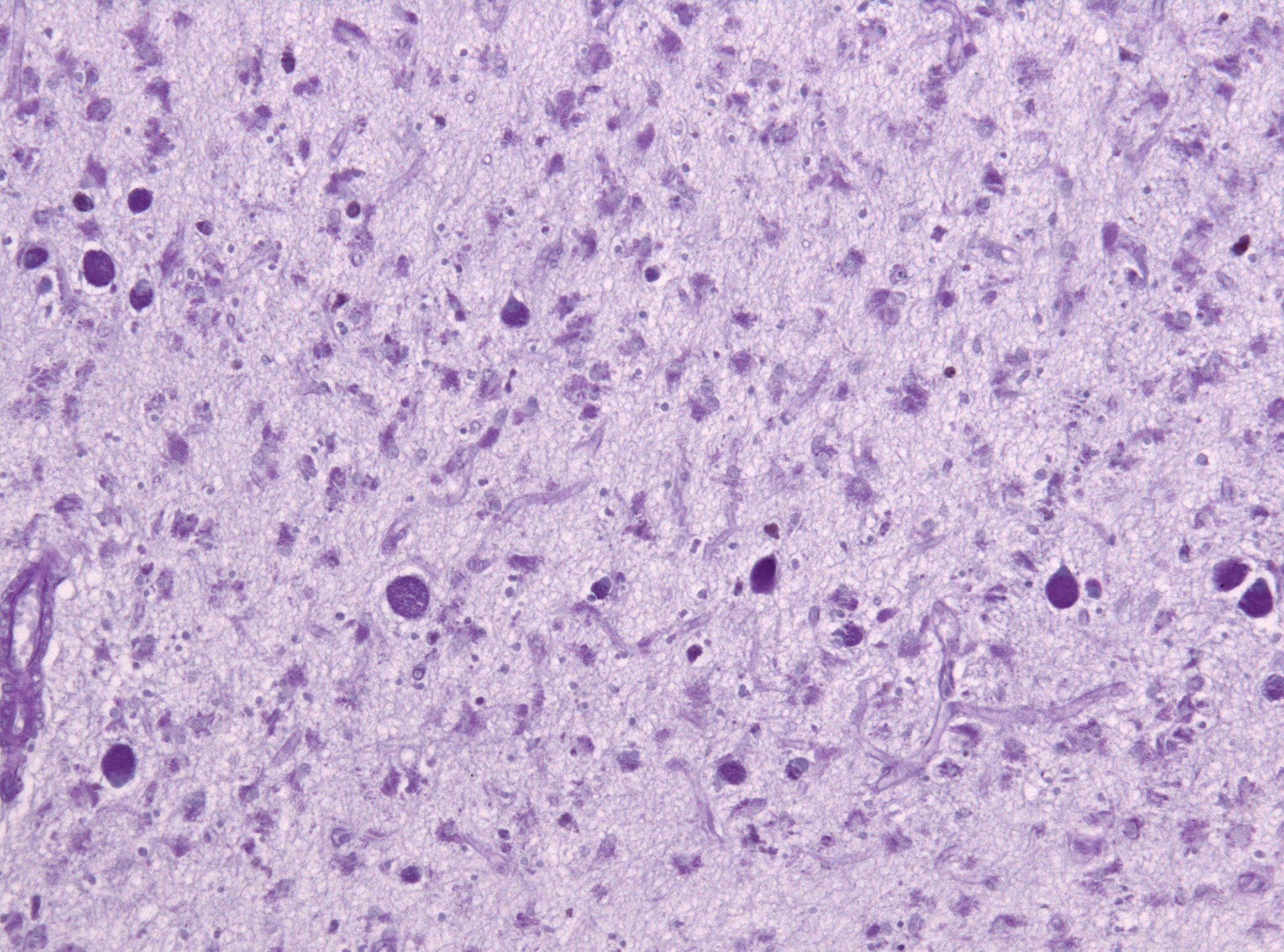
 Histologischer Nachweis der kräftig violett angefärbten Abbauprodukte in den Nervenzellen (PAS)
Histologischer Nachweis der kräftig violett angefärbten Abbauprodukte in den Nervenzellen (PAS)
Die Pathophysiologie der NCL ist noch weitgehend unbekannt. Gesichert ist, dass bei allen Formen die wachsartigen Ceroid-Lipofuszine - aus Fetten und Proteinen bestehende Abfallstoffe des Zellstoffwechsels - intrazellulär im Gewebe gespeichert werden. Dadurch wird das Zellklima toxisch, was zum Absterben der gesunden Zellen führt.
Anfangs äußert sich die Krankheit durch zunehmende Sehschwäche, die schließlich zur vollständigen Erblindung durch Schädigung der Netzhaut (Retinopathie) führt. Damit einhergehend treten bei den Betroffenen Halluzinationen, Epilepsie und Demenz auf. Letztendlich verliert der Patient sämtliche kognitiven und motorischen Fähigkeiten. Da jede NCL-Form unweigerlich zum Tod führt, sind lediglich palliative Behandlungsmethoden möglich.
Klassifizierung
Früher richtete sich die Klassifizierung der NCL-Typen nach dem Manifestationsalter, entsprechend wurden Bezeichnungen wie infantile, spätinfantile oder juvenile NCL verwendet. Die moderne Klassifikation erfolgt auf genetischer Grundlage, die Nummerierung der einzelnen Typen (CLN1, CLN2 usw.) erfolgt dabei nach der historischen Reihenfolge der Entdeckung.
Typ Bezeichnung Manifestationsalter andere Bezeichnung(en) CLN1 infantile NCL spätes Säuglingsalter, auch Erwachsenenalter Hagberg-Santavuori-Krankheit CLN2 (klassische) spätinfantile NCL Kleinkindalter Jansky-Bielschowsky-Krankheit CLN3 (klassische) juvenile NCL Schulalter Stengelsche Krankheit, Vogt-Spielmeyer-Stock-Krankheit (VSS), Batten disease CLN4 adulte NCL (autosomal rezessiv oder dominant) Erwachsenenalter Kufs-Syndrom, Batten-Kufs-Syndrom CLN5 (finnische) spätinfantile NCL Kleinkindalter CLN6 Indisch-iberische NCL Kleinkindalter CLN7 türkische NCL Kleinkindalter CLN8 nordische Epilepsie Schulalter CLN9 noch unbenannt Schulalter Literatur
- Kallenbach, Kurt (Hrsg): Kinder mit besonderen Bedürfnissen. Ausgewählte Krankheitsbilder und Behinderungsformen (ISBN 3-89166-208-4)
- Alfried Kohlschütter, Hans-Hilmar Goebel, Angela Schulz, Zoltan Lukacs: Die neuronalen Ceroid-Lipofuszinosen. Demenzerkrankungen bei Kindern und Jugendlichen in: Deutsches Ärzteblatt, Heft 5, Februar 2005
Siehe auch
Weblinks

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.