- Ostitis deformans
-
Klassifikation nach ICD-10 M88.- Osteodystrophia deformans M88.0 Osteodystrophia deformans der Schädelknochen M88.8 Osteodystrophia deformans sonstiger Knochen M88.9 Osteodystrophia deformans, nicht näher bezeichnet C41.9 (Zusatz bei Kombination mit Osteosarkom) ICD-10 online (WHO-Version 2006) Die Osteodystrophia deformans, auch als Paget-Syndrom, Paget-Krankheit oder Morbus Paget bezeichnet, ist eine Erkrankung des Skelettsystems, bei der es allmählich zu einer Verdickung mehrerer Knochen, meist Wirbelsäule, Becken, Extremitäten oder Schädel kommt. Es handelt sich um eine chronische, langsam fortschreitende Krankheit, an der hauptsächlich ältere Menschen leiden. Sie kann sich auf eine oder zwei Körperstellen beschränken oder sich ausbreiten. Kennzeichnend ist ein rascher Verfall und Umbau der Knochen. Am Beginn der Krankheitsentwicklung steht eine gesteigerte Aktivität der Osteoklasten, welche Knochensubstanz abbauen. Reaktiv folgen dann ungeordnete Anbauvorgänge, wobei die neue Knochenmasse verformt und brüchig ist. Die Krankheitsursache ist nicht sicher bekannt; neuere Forschungen weisen auf genetische Ursachen oder eine Virusinfektion hin.
Inhaltsverzeichnis
Epidemiologie
Die Osteodystrophia deformans tritt häufig in Europa, Australien und Neuseeland auf. Erstbeschreiber und Namensgeber der Krankheit war 1877 der Pathologe und Chirurg James Paget aus England, wo die Krankheit am häufigsten ist. In Westeuropa insgesamt sind etwa 3-4 % der Bevölkerung betroffen; Männer häufiger als Frauen. Allerdings ist nur ein Bruchteil dieser Patienten klinisch auffällig und behandlungsbedürftig. In Asien und Afrika (mit der Ausnahme von Südafrika) ist Osteodystrophia deformans sehr selten.
Ätiologie
Der Entstehungsmechanismus der Osteodystrophia deformans ist unbekannt. Da man paramyxovirale RNA, Antigene und Nukleokapside (z. B. Masernviren) in Osteoklasten, Osteoblasten und Osteozyten findet, geht man von einem viralen Einfluss aus. In Osteoblasten, später Osteoklasten, fördern sie die Bildung des osteoklastenaktivierenden Zytokins Interleukin 6. Auch wird das c-fos-Onkogen exprimiert. Dies erklärt den gelegentlichen späteren Übergang in ein Osteosarkom. Die familiären und geographischen Häufungen sprechen zudem für genetische Einflüsse. Außerdem findet man bei etwa 30% der Patienten das RANK-Gen mutiert (Genlocus 18q21). Dieses exprimiert den Rezeptor-Aktivator für NF-κB, den Regulator der Osteoklastenbildung.
Pathogenese
Die Osteodystrophia deformans tritt in der Regel erst jenseits des 40. Lebensjahres auf, verläuft oft symptomlos und wird im Allgemeinen bei einer Untersuchung oder beim Röntgen wegen anderer Beschwerden festgestellt. Sie beginnt als entzündlicher Prozess in einem oder (meistens) mehreren Knochen und ist zu diesem Zeitpunkt schmerzhaft; die Szintigrafie ergibt eine erhöhte Knochenumbaurate. Röntgenbilder des Knochens zeigen unscharfe Aufhellungen.
Im weiteren Verlauf geht die Entzündung zurück und hinterlässt eine dichte aber unregelmäßige Sklerosierung (kalkreiche Verdichtung) des Knochens, oft auch Deformierungen, druckbedingte Verbiegungen und Auftreibungen der befallenen Skelettelemente, wie eine Wirbelsäulenverkrümmung, einen gewölbter Brustkorb und eine Krümmung der Beine. Bei einer Verdickung der Knochen der Lendenwirbelsäule können Ischiasschmerzen auftreten, die bis ins Bein ausstrahlen (Wurzelkompressionssyndrom).
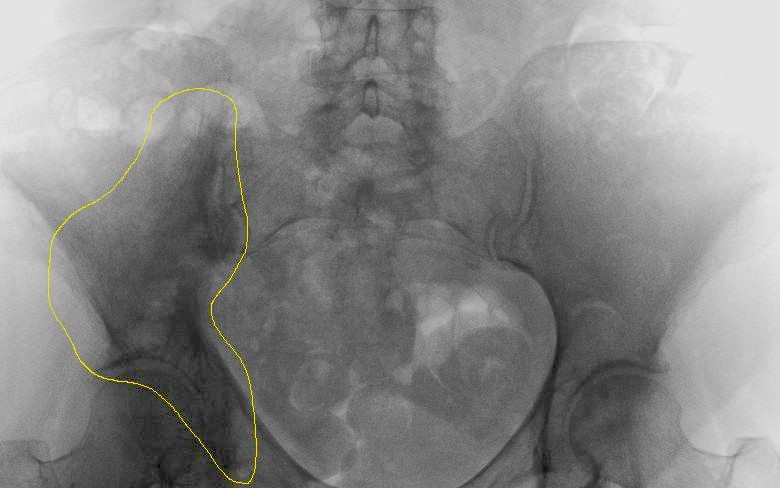
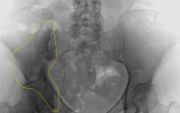 M.Paget im Beckenknochen. Die unscharfe Verdichtung des rechten Darmbeines (im Bild links) ist markiert
M.Paget im Beckenknochen. Die unscharfe Verdichtung des rechten Darmbeines (im Bild links) ist markiertDie Krankheit kann durch lokale Knochenschmerzen auffallen, wobei man durch die Haut die Überwärmung aufgrund der Überaktivität spüren kann. Im fortgeschrittenen Stadium kann es zu einer Verdickung des Schädels mit Zunahme des Kopfumfangs kommen. Sind die Hirnnerven durch das Knochenwachstum geschädigt, kann dies zur Schwerhörigkeit (Versteifung der Gehörknöchelchen, Zuwachsen der Cochlea oder Quetschung des Hörnerven) und Erblindung führen. Feingeweblich ist zu diesem Zeitpunkt ein mosaikähnlich grobmaschiger, aber stabiler lamellärer Reparaturknochen charakteristisch.
Die übermäßige Ausscheidung von Calcium kann zu Nierensteinen führen, die vermehrte Knochendurchblutung begünstigt eine Herzinsuffizienz. Durch die übermäßige Teilungsaktivität der Knochenzellen kommt es in etwa 1% der Fälle zur Entwicklung eines bösartigen Knochentumors. Wenn derartige Komplikationen vermutet werden, ist eine Computertomografie oder Kernspintomografie sinnvoll.
Diagnose
Bildgebende Verfahren
Wesentlich für die Diagnose ist das Röntgenbild, in dem schon im Frühstadium der Erkrankung die Osteolyse nachgewiesen werden kann. Der erhöhte Knochenumbau kann mittels Knochenszintigraphie nachgewiesen werden.
Laboruntersuchungen
Als Ausdruck eines erhöhten Knochenabbaus sind freigesetzte Aminosäuren (vor allem Hydroxyprolin) im Urin nachweisbar. Bedingt durch die vermehrte Aktivität der Osteoblasten ist im Blut ein Anstieg der alkalischen Phosphatase zu verzeichnen, während die Kalziumwerte im Serum normal bleiben. Marker die eine erhöhte Knochenresorption (Abbau) des Typ 1 Collagen beim Knochen anzeigen, wie das C-terminale Telopeptid (CTX), können auch erhöht sein.
Pathologie
Ist nach den oben angeführten Untersuchungen die Diagnose immer noch unklar oder soll ein sekundäres Sarkom ausgeschlossen werden, kann eine Knochenbiopsie durchgeführt werden.
Therapie
Die Behandlung ist symptomatisch mit schmerzlindernden und entzündungshemmenden Medikamenten wie Nichtsteoridalen Antirheumatika, Entlastung des Knochens, Krankengymnastik und gegebenenfalls operativer Stabilisierung von Knochenbrüchen. Die Prognose ist sehr gut. Tritt keine Besserung der Symptome ein, können Medikamente verschrieben werden, die den Verlust der Knochemasse verhindern und Schmerzen lindern; Bisphosphonate und Calcitonin hemmen den Knochenabbau und können bei rechtzeitiger, regelmäßiger Einnahme Deformierungen verhindern. Neuerdings steht auch ein Bisphosphonat zur intravenösen Infusion zur Verfügung. Die einmalige Infusion hat eine Wirkungsdauer von über einem Jahr und ersetzt die tägliche Tabletteneinnahme. Ergänzend werden Vitamin D und Calcium verschrieben. Bei einer besonders schweren Schädigung der Hüfte kann ein Hüftgelenkersatz erforderlich sein.
Literatur
- Piper, Wolfgang: Innere Medizin. Springer Medizin Verlag. Heidelberg 2009.
- Roodman GD, Windle JJ: Paget disease of bone. J Clin Invest. 2005 Feb;115(2):200-8. Review. PMID 15690073
- Walsh JP: Paget's disease of bone. Med J Aust. 2004 Sep 6;181(5):262-5. Review. PMID 15347275
- Griz L, Caldas G, Bandeira C, Assunção V, Bandeira F: Paget's disease of bone. Arq Bras Endocrinol Metabol. 2006 Aug;50(4):814-22. Review. PMID 17117306

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.