- VB-Theorie
-
Die Valenzstruktur-Theorie, auch VB-Theorie, Elektronenpaar-Theorie oder engl. Valence-Bond-Theory genannt, ist ein von Walter Heitler und Fritz London 1927 entwickeltes quantenmechanisches Näherungsverfahren zur Beschreibung von Atombindungen in mehratomigen Systemen.
In diesem Modell entsteht eine Valenzbindung zwischen zwei Atomen dadurch, dass jedes dieser Atome ein Elektron für eine Bindung zur Verfügung stellt. Diese beiden Elektronen bilden ein Elektronenpaar in einem bindenden Molekülorbital. Die beiden Atome im Molekül haben nun Anteil an beiden Elektronen, also am „eigenen“ Elektron und am Elektron „des Partners“. Der energetische Vorteil, der dadurch für die beiden Atome entsteht, ist die Triebkraft für die Bildung von Molekülen.
Inhaltsverzeichnis
Grundmodell
Das Grundmodell wurde für das Wasserstoffmolekül entwickelt, da es die einfachsten Berechnungen ermöglicht:
- Jedes der zwei Wasserstoffatome stellt jeweils ein Elektron für eine Elektronenpaarbindung zur Verfügung.
- Durch Kombination der s-Orbitale der Wasserstoffatome, in denen sich die Elektronen ursprünglich befanden, entstehen Molekülorbitale, ein leeres antibindendes Molekülorbital und ein besetztes bindendes Molekülorbital, in dem sich die beiden Elektronen dann spingekoppelt als Elektronenpaar befinden (Pauliprinzip).
- Die theoretische Energie der Bindung und die Aufenthaltswahrscheinlichkeit der Elektronen im bindenden Molekülorbital lassen sich über die Wellenfunktion dieses Orbitals bestimmen.
- Die Wellenfunktion des bindenden Molekülorbitals jedoch ist unbekannt und wird unter Berücksichtigung verschiedener Faktoren angenähert, bis sie in zufriedenstellender Weise mit experimentellen Befunden übereinstimmt.
- Als Ausgangspunkt für die rechnerische Annäherung dienen die zwei s-Orbitale der ursprünglich einzelnen Wasserstoffatome.
Valenzstruktur-Methode für das Wasserstoff-Molekül
Wasserstoffatom A hat Elektron Nr. 1 und die Wellenfunktion ΨA(1).
Wasserstoffatom B hat Elektron Nr. 2 und die Wellenfunktion ΨB(2).
Der experimentell ermittelte Abstand der Wasserstoffkerne im Molekül liegt bei 74 pm, die Bindungsenergie bei −458 kJ·mol-1.
Erste Näherung
In der ersten Näherung wird ganz außer Acht gelassen, ob und wie sich die beiden Atomkerne und Elektronen gegenseitig beeinflussen, wenn sie sich zur Bildung einer Bindung einander annähern. Die Wellenfunktion Ψ für ein System aus zwei Atomen, die sich gegenseitig nicht beeinflussen, erhält man aus den Wellenfunktionen der einzelnen Wasserstoffatome:
Die Bindungsenergie und der Kernabstand, die sich daraus ergeben, stimmen kaum mit den experimentellen Befunden überein.
Austauschenergie nach Heitler und London
Im Molekülorbital muss sich Elektron 1 nicht immer bei Wasserstoffatom A befinden, genauso wenig Elektron 2 immer bei Wasserstoffatom B. Dementsprechend wird ein Term für vertauschte Elektronen hinzu gefügt:
Hier findet man schon eine gute Annäherung an experimentelle Ergebnisse.
Abschirmung
Die Terme ΨA und ΨB berücksichtigen nur, dass ein Elektron die Kernladung eines Wasserstoffkernes abschirmt. Im Molekül jedoch befinden sich zwei Kerne und zwei Elektronen dazwischen, sodass der Abschirmungseffekt größer ist.
Die Berücksichtigung der effektiven Kernladung findet in den Termen für Ψ'A und Ψ'B statt, die obige Gleichung kann also prinzipiell so stehen bleiben:
Resonanz
Theoretisch können die Wasserstoffatome im Verbund ihre Elektronen nicht nur austauschen, sondern es besteht auch die kleine Wahrscheinlichkeit, dass sich manchmal beide Elektronen an einem der Wasserstoffkerne befinden. Dementsprechend lassen sich ionische Strukturen für das Wasserstoffmolekül H-H formulieren:
Da die ionischen Strukturen aber wahrscheinlich selten auftreten, wird ihr Einfluss auf die Wellenfunktion Ψ mit einem Faktor λ < 1 versehen:
Hier ist die Abweichung von experimentellen Befunden schon gering und nach Anwendung einer Wellengleichung mit 100 Korrekturtermen kommt man zu Ergebnissen, die mit den Experimentbefunden fast übereinstimmen.
Das Grundmodell für Wasserstoffmoleküle wurde immer weiter verfeinert und die Problematik auf größere und wesentlich kompliziertere Moleküle übertragen, sowie auf Mehrfachbindungen. Die Herangehensweise der Valenzstrukturmethode, sowie die Molekülorbitaltheorie stellen die Grundlage des heutigen Molecular Modelling dar, das durch computergestützte Berechnungen Voraussagen und Deutungen vieler Molekülstrukturen und Eigenschaften ermöglicht.
Paulingsche Theorie der Komplexe
In Komplexen aus Zentralatom und einer bestimmten Anzahl von Liganden liegt die koordinative Bindung vor. Diese Bindungsart kommt nicht dadurch zustande, dass beide Reaktionspartner, also Zentralatom und Ligand, jeweils ein Elektron zur Verfügung stellen, sondern dadurch, dass der Ligand alleine zwei Elektronen mitbringt und damit eine Bindung zum Zentralatom ausbildet.
Wenn ein Ligand zwei Elektronen liefert, und n die Anzahl der bindenden Liganden ist, dann bekommt das Zentralatom n×2 Elektronen, die es irgendwo unterbringen muss. Zur Unterbringung stehen die leeren Außenorbitale des Zentralatoms zur Verfügung:
- Erste Periode der Übergangsmetalle: (außen) 4 d, 4 p, 4 s, 3 d (innen)
- Zweite Periode der Übergangsmetalle: (außen) 5 d, 5 p, 5 s, 4 d (innen)
- Dritte Periode der Übergangsmetalle: (außen) 6 d, 6 p, 6 s und 5 d (innen)
inner/outer orbital Komplexe
Die Aufklärung, warum manche Liganden outer- oder inner sphere Komplexe erzeugen, gelang erst mit der Kristallfeld- bzw. Ligandenfeldtheorie. Hier wurde der Begriff von high spin und low spin Komplexen eingeführt entsprechend der magnetischen Eigenschaften solcher Komplexe.
Beispiel:
Das Fe2+-Kation hat 6 Elektronen im 3d-Orbital, also eine 3d6-Konfiguration.
Mit sechs Wasserliganden kommen 12 Elektronen hinzu. In diesem Fall bleibt die Konfiguration des Kations erhalten: 3d6 4s2 4p6 4d4.
Mit sechs Cyanidliganden kommen auch 12 Elektronen hinzu. Hier verändert sich die "Originalbesetzung" des Zentralatoms zu: 3d10 4s2 4p6 5s2.
Geometrie
Zur Bindungsbildung zwischen Zentralatom und Liganden werden nach der VB-Theorie Hybridorbitale von Seiten des Zentralatoms gebildet, deren Anzahl der Menge an Elektronenpaaren entspricht, die die Liganden zur Bindung zur Verfügung stellen.
Je nach Art und vor allem Anzahl der Liganden-Elektronenpaare werden zur Hybridisierung bestimmte d(innen)-, s-, p- und d(außen)-Orbitale des Zentralatoms benutzt, wodurch sich charakteristische Koordinationsgeometrien ergeben:
- 6 Liganden: 2 · d (innen) + 1 · s + 3 · p = 6 · d2sp3 --> Oktaeder (inner orbital complex)
- 6 Liganden: 1 · s + 3 · p + 2 · d (außen) = 6 · sp3 d2 --> Oktaeder (outer orbital complex)
- 4 Liganden: 1 · s + 3 · p = 4 · sp3 --> Tetraeder
- 4 Liganden: 1 · d(innen) + 1 · s + 2 · p = 4 · dsp2 --> planares Quadrat
usw.
Grenzen
Die VB-Theorie eignet sich gut zur Bestimmung von Komplexgeometrien und zur Erklärung der magnetischen Phänomene. Andere Phänomene, wie die Farbigkeit von Komplexen, lassen sich jedoch mit der VB-Theorie nicht erklären. Dies gelingt mit weiterführenden Theorien wie der Kristallfeldtheorie bzw. Ligandenfeldtheorie und der Molekülorbital-Theorie.
Literatur
- Charles A. Coulson: Die chemische Bindung. Hirzel, 1969.
- James E. Huheey, Ellen Keiter, Richard L. Keiter: Anorganische Chemie. Prinzipien von Struktur und Reaktivität. 3. Auflage. Gruyter, 2003, ISBN 3110179032.
- Nils Wiberg, Egon Wiberg, Arnold Fr. Holleman: Lehrbuch der Anorganischen Chemie. 101. Auflage. Gruyter, 1995, ISBN 3110126419.
Siehe auch
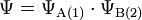
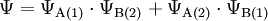
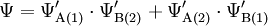
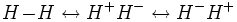
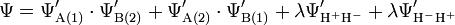
Wikimedia Foundation.