- Molekülorbital-Theorie
-
Die Molekülorbitaltheorie (kurz MO-Theorie) ist eine von zwei komplementären Möglichkeiten den Aufbau von Atombindungen zu beschreiben, die andere Möglichkeit ist die VB-Theorie. Beim MO-Verfahren werden die Atomorbitale der beteiligten Atome ‚vermischt‘. Dabei spalten sie sich in bindende und antibindene Molekülorbitale auf.
Das VB-Verfahren geht von lokalisierten Bindungen aus (siehe Valenzstrukturtheorie). Mit diesem von Walter Heitler und Fritz London 1927 entwickelten Verfahren konnte das H2-Molekül näherungsweise berechnet werden, was als Begründung der Quantenchemie gesehen werden kann. Etwas später wurde von Friedrich Hund und Robert S. Mulliken das MO-Verfahren entwickelt, das heute für die meisten quantenchemischen Rechnungen verwendet wird.
Inhaltsverzeichnis
Physikalische Erklärung
Eine n-Elektronen-Wellenfunktion hat, wenn der Spin nicht beachtet wird, die allgemeine Form
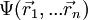 . Der Betrag des Produktes mit der komplex konjugierten Funktion
. Der Betrag des Produktes mit der komplex konjugierten Funktion 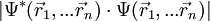 gibt die Wahrscheinlichkeitsdichte wieder, das erste Elektron an der Stelle
gibt die Wahrscheinlichkeitsdichte wieder, das erste Elektron an der Stelle 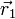 , das 2-te an der Stelle
, das 2-te an der Stelle 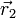 usw. zu finden.
usw. zu finden.Die wahre Wellenfunktion lässt sich analytisch nicht finden. Eine zielführende Vereinfachung ist es, die Elektronen als statistisch unabhängig anzusehen. Mathematisch bedeutet das, einen Produktansatz zu verwenden
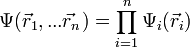 . Dieser Ansatz ist auch als Hartree-Produkt bekannt. Die
. Dieser Ansatz ist auch als Hartree-Produkt bekannt. Die 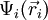 geben die Aufenthaltsbereiche für die einzelnen Elektronen an. Sie werden als Molekülorbitale bezeichnet. Um das Pauli-Prinzip einzuhalten, wird die Wellenfunktion als Slater-Determinante (einer Summe von n Produkten) angesetzt. Dann sind die Elektronen ununterscheidbar und wechseln zwischen allen Orbitalen.
geben die Aufenthaltsbereiche für die einzelnen Elektronen an. Sie werden als Molekülorbitale bezeichnet. Um das Pauli-Prinzip einzuhalten, wird die Wellenfunktion als Slater-Determinante (einer Summe von n Produkten) angesetzt. Dann sind die Elektronen ununterscheidbar und wechseln zwischen allen Orbitalen.Abgesehen davon, dass MO-Schemata im allgemeinen nicht die wahre Situation wiedergeben können, ist zu beachten, dass sie in der MO-Theorie nicht eindeutig bestimmt sind. Entscheidend ist nur die Summe über alle quadrierten Orbitale, die Elektronendichte (das ist auch die Grundlage für die Dichtefunktionaltheorie). Mathematisch gesprochen ist die Wellenfunktion gegenüber einer unitären Lineartransformation invariant. Ein Beispiel dazu sind die beiden angegebenen Modelle zur Beschreibung der Doppelbindung.
Mathematische Grundlagen
Gesucht werden Lösungen der Schrödingergleichung eines Moleküls. Die Rechnungen sind aber wesentlich schwieriger auszuführen als bei einem isolierten Atom. Im Normalfall, wenn mehr als ein Elektron betrachtet wird, gibt es im Sinne eines Dreikörperproblems keine analytisch angebbaren exakten Lösungen. Daher müssen Näherungsmethoden herangezogen werden. Dafür eignen sich das VB-Verfahren und das MO-Verfahren, die zu ähnlichen Ergebnissen führen.
Zur näherungsweisen Bestimmung der Molekülorbitale dient die Variationsrechnung. Diese basiert auf der Tatsache, dass wegen der Hermitezität des Hamilton-Operators der Energieerwartungswert jeder beliebigen Funktion höher als der tiefste Energieeigenwert ist. Man muss also in einer Extremwertaufgabe die Funktion mit dem tiefsten Energieerwartungswert auswählen. Diese ist dann wahrscheinlich die beste Näherung.
Es reicht aber nicht aus, eine einmalige Variationsrechnung durchzuführen. Über die Hartree-Fock-self-consistent-field-Methode werden Orbitale iterativ bestimmt. Das Prinzip ist, dass für jedes Elektron, das Feld der anderen Elektronen berücksichtigt wird, welches allerdings nicht exakt bekannt ist, sondern im Verlauf der Iteration immer besser wird.
Wichtig ist auch die Born-Oppenheimer-Näherung, nach der die Elektronen- und Kernbewegung isoliert betrachtet werden können. Somit können Elektronenverteilung und Schwingung getrennt behandelt werden.
VB-Verfahren
Die Valenzstrukturtheorie (VB-Theorie oder VB-Verfahren von engl. valence bond) nach Walter Heitler, Fritz London, John C. Slater und Linus Carl Pauling geht von lokalisierten Bindungen aus. Sie wird durch die üblichen Strukturformeln (u.U. mit Mesomere) repräsentiert. Sie war historisch entscheidend für das Verständnis der chemischen Bindungen. Heutige Rechnungen werden eher mit der MO-Methode durchgeführt.
Rechnerisch werden die Elektronen in verschiedener Art auf die Atomorbitale verteilt und die Linearkombination dieser Valenzstrukturen gebildet, die die geringste Energie besitzt und daher gemäß dem Variationsprinzip die beste Näherung ist.
Ein entscheidendes Prinzip bei der VB-Theorie ist Promotion der Elektronen und Hybridisierung der Orbitale.
MO-Verfahren
Das MO-Verfahren (von engl. molecular orbital) nach Friedrich Hund und Robert Sanderson Mulliken ordnet alle Elektronen des Moleküls einem Satz Molekülorbitalen zu. Die Veranschaulichung erfolgt durch Elektronenwolken, die meist über das gesamte Molekül delokalisiert sind.
Molekülorbitale können als Linearkombinationen zu einer endlichen Basis angesetzt werden. Dann werden in einem erweiterten Eigenwertproblem die Molekülorbitale bestimmt. Als Basis können, wie von Lennard-Jones vorgeschlagen, die Atomorbitale der isolierten Atome im Sinne der LCAO-Näherung (Linear Combination of Atomic Orbitals) verwendet werden.
Grundsätzlich könnten beliebige Funktionen als Basis herangezogen werden. Gute Lösungen mit wenig Rechenaufwand werden erhalten, wenn physikalisch sinnvolle Funktionen verwendet werden. Dafür eignen sich, wie Lennard-Jones als erster feststellte, die Atomorbitale, die in isolierten Atomen die Elektronen richtig beschreiben. Man spricht dann von LCAO. Zur Verbesserung können auch die Atomorbitale variiert werden oder weitere Funktionen in den Basissatz eingeschlossen werden.
Das MO-Verfahren kann bei kleinen symmetrischen Molekülen intuitiv verstanden werden. Aus Symmetriegründen ergeben sich die Molekülorbitale aus Addition bzw. Subtraktion der Atomorbitale. Bei komplizierteren Molekülen setzen sich die Molekülorbitale als Linearkombination von verschiedenen Atomorbitalen zusammen. Genau genommen wechselwirken auch schon in der zweiten Periode die 2s und 2pz-Orbitale, sodass auch schon dort kompliziertere Linearkombinationen erhalten würden. Bei konjugierten π-Systemen stellt die Hückel-Näherung eine Methode zur groben Bestimmung von MOs dar.
Ein grundsätzlicher Fehler dieser Methode ist, dass die Elektronen (bis auf Einhaltung des Pauli Prinzips) als statistisch unabhängig voneinander gesehen werden. Viel aufwendigere korrelierte Rechnungen, v.a. CI (configuration interaction), beachten auch die Elektronen-Korrelation.
Zeichnen von LCAO-MO-Diagrammen
Qualitative LCAO-MO-Diagramme können auch ohne Rechnung gezeichnet werden. Zu beachten ist, dass bei der Linearkombination zweier AOs ein bindendes MO mit tieferer Energie als das tieferliegende AO und ein antibindendes MO mit höherliegender Energie als das höherliegende AO gebildet werden. Die Aufspaltung wird in erster Näherung von der Überlappung bestimmt. So kann man z. B. vorhersagen, dass eine σ-Bindung stärker aufspaltet als eine π-Bindung.
σ-Bindung
σ-Bindung 
Molekülorbital von H2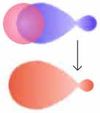
s-q-MO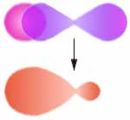
s-p-MO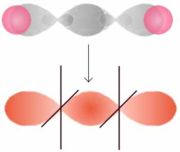
q-q-MOAls σ-Bindung wird eine Bindung bezeichnet, die rotationssymmetrisch zur Bindungsachse ist. Anders ausgedrückt werden Orbitale mit der magnetischen Quantenzahl ml = 0 kombiniert, d. h. s-, px-, dz2-Orbitale und Mischungen (Hybride) aus diesen.
Beispiele:
- Das Wasserstoff-Molekülorbital entsteht durch Überlappung der 1s-Orbitale der Wasserstoffatome. Die kleinen Kreise entsprechen dem Bindungsabstand, die großen Kreise dem Atomradius.
- Im Wassermolekül verbinden sich die 1s-Orbitale von zwei Wasserstoffatomen mit je einem sp3-Hybridorbital des Sauerstoff-Atoms zu zwei σ-Bindungen. Die vier Orbitale der bindenden und nichtbindenden Elektronenpaare sind nach den Ecken eines Tetraeders ausgerichtet. Die rot eingezeichneten Elektronenpaare befinden sich in den Atomorbitalen und die grauen Elektronenpaare in den Molekülorbitalen.
- Im Fluorwasserstoff verbindet sich das kugelige 1s-Orbital des Wasserstoffatoms mit dem hantelförmigen px-Orbital des Fluoratoms zu einem Molekülorbital mit ungleichen Orbitalhälften. (Die nichtbindenden py- und pz-Orbitale sind nicht eingezeichnet.)
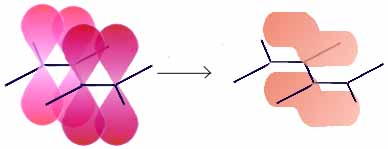
- Im Ethin verbinden sich zwei sp-Hybridorbitale der Kohlenstoffatome zu einem Molekülorbital, die anderen Hybridorbitale bilden mit den 1s-Orbitalen der Wasserstoffatome ebenfalls je ein Molekülorbital, die py- und pz-Orbitale der C-Atome, die nicht zur Hybridisierung der Atomorbitale benutzt wurden, stehen senkrecht zur Bindungsachse und bilden zwei π-Bindungen (siehe unten). (In der Abbildung sind zusätzlich zu den Molekülorbitalen die p-Orbitale als schwarze Linien angedeutet)
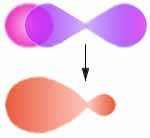
Das Molekülorbital der Doppelbindung
Es ist spiegelsymmetrisch bezüglich der Bindungsachse.
σ-π-Modell
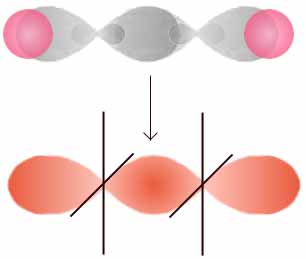
π-Bindung 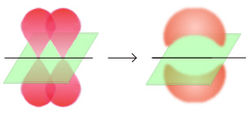
Molekülorbital der π-Bindung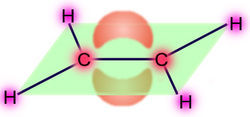
Molekülorbital der π-Elektronen im EthenEine Doppelbindung besteht aus einer σ-Bindung und aus einer π-Bindung, wobei die Bindungspartner im sp2-hybridisierten Zustand vorliegen: drei Hybridorbitale weisen in die Ecken eines gleichseitigen Dreiecks, senkrecht dazu steht das pz-Orbital, das nicht für die Hybridisierung verwendet wurde. Die σ-Bindung entsteht durch Überlappung zweier Hybridorbitale, die π-Bindung entsteht durch Überlappung der zwei pz-Orbitale. Da beide pz-Orbitale parallel zu einander stehen müssen, entsteht ein neues Molekülorbital mit einer Knotenebene.
Das π-Molekülorbital wird durch Kombination von Orbitalen mit |ml| = 1 gebildet. Es enthält eine Knotenebene in der Kernachse.
- Beispiel Ethen: Die beiden Hälften des π-Molekülorbitals liegen ober- und unterhalb der Ebene der σ-Bindungen (grün, die C-C- und H-C-Sigma-Bindungen sind nur als schwarze Linien dargestellt).
- Beispiel Ethin: Die Bindungssituation im Ethin (Trivialname Acetylen), das eine Dreifachbindung enthält, setzt sich aus einer σ-Bindung, die zwischen der Kernverbindungsachse lokalisiert ist, und zwei π-Bindungen zusammen.
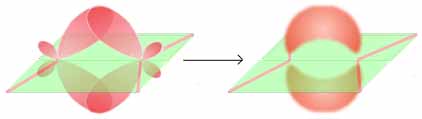
τ-Modell
τ-Modell 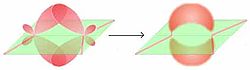
Molekülorbitale nach dem τ-ModellEine selten angewandte Methode zur Beschreibung von Doppelbindungen ist das τ-Modell. s- und p-Orbitale werden zuerst gemischt (beide C-Atome sind sp³-hybridisiert) und aus den beiden Hybridorbitalen die Doppelbindung zusammengesetzt. Die τ-Bindungen entstehen durch Überlappung von jeweils zwei Hybridorbitalen, es bilden sich zwei spiegelbildliche Molekülorbitale („Bananen-Bindungen“). Es zeigt sich, dass das τ-Modell Bindungswinkel und -längen passend wiedergibt.
Die Unterscheidung ist nur in der VB-Theorie sinnvoll. Bei einer LCAO-Methode gehen die beiden Modelle ineinander über, da in beiden Fällen in der Summe die gleiche Elektronendichte erhalten wird. Diese ist das einzig relevante.
Delokalisation
Konjugierte π-Bindung 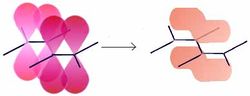
Delokalisiertes Molekülorbital bei 1,3-ButadienDelokalisation tritt dann auf, wenn ein Molekül mehrere Doppelbindungen enthält, die konjugiert sind. Das heißt, dass zwischen ihnen immer genau eine Einfachbindung ist. Dazu müssen alle pz-Orbitale zueinander parallel und in direkter Nachbarschaft stehen. Dann können alle pz-Orbitale zu einem einzigen Molekülorbital kombiniert werden, was quantenmechanisch bewiesen werden kann.
Beispiele in chemischen Verbindungen
Wasserstoff
Wasserstoff bindend antibindend 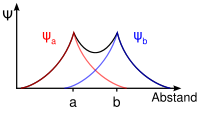
Additive Überlagerung der Wellenfunktion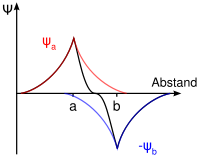
Subtraktive Überlagerung der Wellenfunktion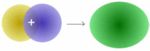
Bindendes Molekülorbital
Antibindendes MolekülorbitalBesetzung der Molekülorbitale von Wasserstoff und Helium Wasserstoff Helium 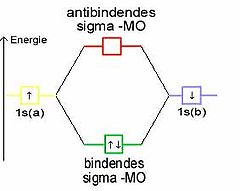
Besetzung beim Wasserstoff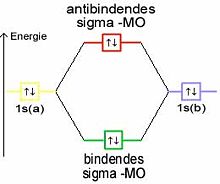
Besetzung beim HeliumDie zur Bindung notwendigen einsamen Elektronen befinden sich jeweils im 1s-Orbital der beiden Atome Ha und Hb, das durch die Eigenfunktionen ψa(1s) und Ψb(1s) beschrieben wird.
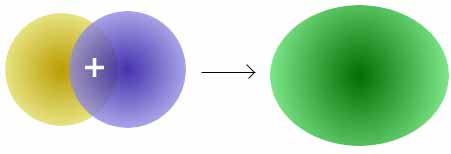
Die Addition der Wellenfunktionen ψa(1s) + ψb(1s) ergibt ein rotationssymmetrisches bindendes Molekülorbital ( σ(1s) ) mit erhöhter Ladungsdichte zwischen den Kernen der Bindungspartner. Durch die Anziehung der Kerne durch die Ladung hält das Molekül zusammen.
Die Subtraktion der Wellenfunktionen ψa(1s) - ψb(1s) ergibt ein antibindendes Molekülorbital ( σ*(1s) ) mit einer Knotenebene zwischen den Kernen der Bindungspartner. Durch die resultierende geringe Elektronendichte zwischen den Kernen kommt es zu einer Abstoßung der Atome.
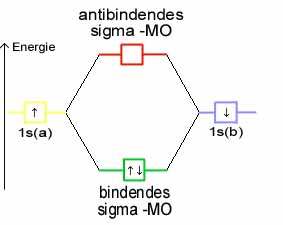
Die Molekülorbitale können (wie die Atomorbitale) mit maximal zwei Elektronen entgegengesetzten Spins besetzt werden. Da jedes Wasserstoffatom jeweils ein Elektron zur Verfügung stellt, wird das bindende Molekülorbital im energieärmsten Grundzustand mit einem Elektronenpaar besetzt, während das antibindende leer bleibt. (Im angeregten Zustand ist das bindende und das antibindende Molekülorbital mit je einem Elektron besetzt.)
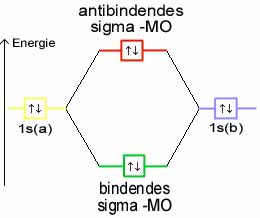
Ein anderes Beispiel ist Helium. Hier ist jedes 1s-Orbital bereits mit einem Elektronenpaar besetzt. Bei der Kombination dieser Atomorbitale müsste sowohl das bindende als auch das antibindende Molekülorbital mit je einem Elektronenpaar besetzt werden. Ihre Wirkungen würden sich gegenseitig aufheben, es kommt keine Bindung zustande.
Sauerstoff
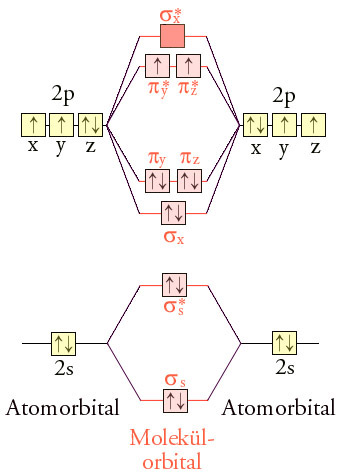
LCAO-MO-Schema von Triplett-Sauerstoff 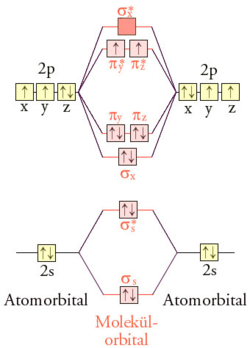
Besetzung der EnergieniveausDas LCAO-MO-Schema kann wie oben beschrieben qualitativ abgeleitet werden. Jedes Sauerstoff-Atom hat im Grundzustand sechs Valenzelektronen auf dem zweiten Hauptenergieniveau. Die zwölf Valenzelektronen eines O2-Sauerstoffmoleküls werden auf die vier bindenden (σs, σx, πy, und πz) und drei der vier antibindenden Molekülorbitale (σs*, πy*, πz*) verteilt. Da zwei antibindende Orbitale mit nur einem Elektron besetzt sind (eine „halbe Bindung“), resultiert eine Doppelbindung.
Di-Sauerstoff hat im Grundzustand, einem Triplettzustand, gemäß der Hund’schen Regel zwei ungepaarte Elektronen parallelen Spins. Durch diese Elektronenverteilung lässt sich der Paramagnetismus und der diradikalische Charakter des Sauerstoffs erklären. Interessanterweise senkt der Diradikalcharakter die Reaktionsfähigkeit, da eine konzertierte Reaktion der Spinerhaltung widersprechen würde. Besonders reaktionsfähig ist der angeregte diamagnetische Singulett-Sauerstoff.
Eine weitere Folge der MO-Besetzung ist, dass es für O2 schwierig ist, eine korrekte Lewis-Formel anzugeben. Entweder wird der Diradikalcharakter vernachlässigt oder die Doppelbindung.
Butadien
π-System des Butadiens bindend antibindend 
stark bindendes MO (besetzt)
stark antibindendes MO (unbesetzt)
schwach bindendes MO (besetzt)
schwach antibindendes MO (unbesetzt)Das π-System des Butadiens setzt sich zusammen aus 4 pz-Orbitalen, die am Anfang mit je einem Elektron besetzt sind. Diese 4 Atomorbitale werden nun zu vier Molekülorbitalen linear kombiniert. Die Koeffizienten erhält man durch Symmetrie-angepasste-Linearkombination (SALC) oder nach der Hückel’schen Theorie. Dabei entstehen die rechts gezeichneten Orbitale. Die rot/blaue Färbung gibt an, ob das Orbital vor dem Quadrieren ein negatives oder positives Vorzeichen hatte. Physikalisch hat sie keine Relevanz.
Jedes dieser Orbitale kann mit 2 Elektronen besetzt werden. Es werden also die beiden unteren Orbitale voll aufgefüllt und die beiden oberen bleiben leer. Energetisch besonders günstig ist das Orbital, bei dem alle pz-Orbitale das gleiche Vorzeichen haben und sich daher die Elektronen fast frei über das ganze Molekül bewegen können.
Man erkennt die von SALC geforderte Eigenschaft, dass in jedem Molekülorbital alle Symmetrieelemente des Moleküls erhalten bleiben. Weiterhin sieht man, wie mit zunehmender Energie die Anzahl an Knotenebenen steigt.
Bindungsordnung
Die Bindungsordnung bezeichnet die Zahl der effektiven Bindungen in einem Molekül. Sie ist die Hälfte der Differenz der bindenden und der antibindenden Valenzelektronen (s. Bindungsordnung). Sie ist einfach abzulesen, da sie gleich der Anzahl der Bindungsstriche in der Lewis- Schreibweise der Verbindung ist.
Weblinks
Siehe auch









Wikimedia Foundation.