- Bernard-Soulier-Syndrom
-
Klassifikation nach ICD-10 D69.1 Thrombozytopathie ICD-10 online (WHO-Version 2011) Das Bernard-Soulier-Syndrom (BSS), auch als Hämorrhagische Thrombozytendystrophie bezeichnet, ist eine sehr seltene autosomal-rezessiv vererbte Blutungskrankheit, die zu den Thrombozytopathien gerechnet wird.
Inhaltsverzeichnis
Ursache und Genetik
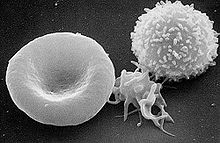 REM-Aufnahme von Blutkörperchen.
REM-Aufnahme von Blutkörperchen.
Von links nach rechts: Erythrozyt (rotes Blutkörperchen), normaler Thrombozyt (aktiviert) und LeukozytDie Blutungen werden durch eine Funktionsstörung der Thrombozyten verursacht, die ein agglutinieren der Thrombozyten verhindert. Ursache der Funktionsstörung ist ein Mangel oder eine Dysfunktion des Glykoprotein-Ib-V-IX-Komplexes (GPIb-V-IX). GPIb-V-IX wird nur von Thrombozyten exprimiert. Es ist ein aus mehreren Untereinheiten zusammengesetzter Rezeptor, der bei primären Hämostase (Blutgerinnung) zur Bindung des von-Willebrand-Faktors (vWF, ein Trägerprotein) von entscheidender Bedeutung ist. Erst durch Bindung von vWF ist die Adhäsion der Thrombozyten an das verletzte Endothel, sowie die des von-Willebrand-Faktors und das Agglutinieren auch unter hohen Scherkräften möglich.[1]
Der GPIb-V-IX-Komplex besteht aus vier Untereinheiten, die durch vier verschiedene Gene kodiert werden:[1]
- GPIb-Alpha durch Chromosom 17 Genlocus 17pter-p12
- GPIb-Beta durch Chromosom 22 Genlocus 22q11.2
- GPV durch Chromosom 3 Genlocus 3q29
- GPIX durch Chromosom 3 Genlocus 3q21
Mit Ausnahme des GPV-Gens wurden in allen Genen Mutationen gefunden, die für die Entstehung des Bernard-Soulier-Syndroms verantwortlich sind. Über 30 verschiedene Mutationen im GPIb-Alpha-, GPIb-Beta- oder GPIX-Gen wurden in Assoziation mit dem Bernard-Soulier-Syndrom beschrieben.[2]
Epidemiologie und Prävalenz
Das Bernard-Soulier-Syndrom ist eine ausgesprochen seltene Erkrankung. Bisher sind weltweit etwa 100 Fälle in der Literatur beschrieben. Die Prävalenz wird auf 0,1:100.000 geschätzt, allerdings liegt sie bedingt durch Fehldiagnosen und Nichterkennung wahrscheinlich höher.[3]
Symptome und Diagnose
Das Bernard-Soulier-Syndrom manifestiert sich durch Blutungsneigung und sogenannte Riesen-Thrombozyten (Makrothrombozyten). Bei einem Teil der Patienten kann zudem die Thrombozytenzahl vermindert sein (Thrombozytopenie).[4] Die Thrombozytenzahl liegt bei BSS-Patienten im Bereich von kleiner 30.000 bis 200.000 pro µl (normal: 150.000 bis 400.000). Der Durchmesser der Thrombozyten liegt bei 4 bis 10 µm (normal: 1 bis 4 µm). Die Blutungsdauer liegt zwischen 5 und mehr als 20 Minuten (normal: 2 bis 7 min).[5][1]
Klinisch kann sich das Bernard-Soulier-Syndrom durch starkes Nasen- und Zahnfleischbluten, gastrointestinale Blutungen, sowie durch Purpura (Kapillarblutungen) manifestieren. Bei Frauen kann es zu einer verlängerten Regelblutung (einer Menorrhagie) beziehungsweise einer Hypermenorrhoe kommen.
Über die verlängerte Blutungszeit an der Haut, dem Auftreten von Riesen-Thrombozyten (Makrothrombozytopenie), dem in-vitro Ausbleiben der Agglutination (Aggregation der Thrombozyten) bei Zusatz von Ristocetin zum Blut, sowie anhand der niedrigen oder fehlenden Expression der GPIb-V-IX-Komplexe, kann eine sichere Diagnose gestellt werden.[6]
Therapie
Die Therapie erfolgt – wenn notwendig – im Wesentlichen symptomatisch. Vor Operationen ist unter Umständen eine Thrombozytentransfusion notwendig.
Prognose
Die Prognose ist in der Regel für die Patienten günstig.
Entdeckung
Das Bernard-Soulier-Syndrom ist nach den beiden französischen Hämatologen Jean Bernard (1907–2006) und Jean-Pierre Soulier (1915–2003) benannt, die dieses Syndrom 1948 als erste beschrieben.[7]
Einzelnachweise
- ↑ a b c F. Lanza: Hemorrhagiparous Thrombocytic Dystrophy. In: Orphanet Encyclopedia Oktober 2003
- ↑ S. Kunishima u. a.: Genetic abnormalities of Bernard-Soulier syndrome. In: Int J Hematol 76/2002, S. 319–27. PMID 12463594
- ↑ V. Sundararajan, G. S. Retzinger: Bernard-Soulier Syndrome. In: LabLine 8/2002
- ↑ J. A. Lopez u. a.: Bernard-Soulier syndrome. In: Blood 91/1998, S. 4397–418. PMID 9616133
- ↑ The University of Chicago Genetic Services Laboratories: GP Ibß analysis for Bernard-Soulier Syndrome
- ↑ F. Lanza: Bernard-Soulier-Syndrom orpha.net
- ↑ J. Bernard, J. P. Soulier: Sur une nouvelle variete de dystrophie thrombocytaire-hemorragipare congenitale. In: Semin Hop Paris 24/1948, S.3217–22. PMID 18116504
Literatur
- F. Lanza: Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). In Orphanet J Rare Dis 16/2006, S. 46. PMID 17109744
- M. L. Budarf u. a.: Identification of a patient with Bernard-Soulier syndromeand a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11.2. In: Hum Mol Genet 4/1995, S. 763–6.
- M. Yagi u. a.: Structural characterization and chromosomal location of the gene encoding human platelet glycoprotein Ibß. In: J Biol Chem 269/1994, S. 17424–7.
- C. Li u. a.: Bernard-Soulier syndrome with severe bleeding: absent platelet glycoprotein Ib alpha due to a homozygous one-base deletion. In: Thromb Haemost 76/1996, S. 670–4. PMID 8950770
- J. Ware u. a.: Generation and rescue of a murine model of platelet dysfunction: the Bernard-Soulier syndrome. In: Proc Natl Acad Sci U S A 97/2000, S. 2803–8. PMID 10706630
- A. Tomer u. a.: Bernard-Soulier syndrome: quantitative characterization of megakaryocytes and platelets by flow cytometric and platelet kinetic measurements. In: Eur J Haematol 52/1994, S. 193–200. PMID 8005229
- C. Li u. a.: The genetic defect in two well-studied cases of Bernard-Soulier syndrome: a point mutation in the fifth leucine-rich repeat of platelet glycoprotein Ib alpha. In: Blood 86/1995, S. 3805–14. PMID 7579348
- J. P. Caen u. a.: Bernard-Soulier syndrome: a new platelet glycoprotein abnormality. Its relationship with platelet adhesion to subendothelium and with the factor VIII von Willebrand protein. In: J. Lab. Clin. Med. 87/1976, S. 587–96.
- C. N. Finch u. a.: Evidence that an abnormality in the glycoprotein Ib alpha gene is not the cause of abnormal platelet function in a family with classic Bernard-Soulier disease. In: Blood 75/1990, S. 2357–62. PMID 1972029
- I. Hagen u. a.: Immunochemical evidence for protein abnormalities in platelets from patients with Glanzmann's thrombasthenia and Bernard-Soulier syndrome. In: J. Clin. Invest. 65/1980, S. 722–31. PMID 7354135
- J. Ware u. a.: Nonsense mutation in the glycoprotein Ib-alpha coding sequence associated with Bernard-Soulier syndrome. In: Proc. Nat. Acad. Sci. 87/1990, S. 2026–30. PMID 2308962
- R. B. Stricker u. a.: Acquired Bernard-Soulier syndrome: evidence for the role of a 210,000-molecular weight protein in the interaction of platelets with von Willebrand factor. In: J. Clin. Invest. 76/1985, S. 1274–8. PMID 2931453
- J. L. Moake u. a.: Binding of radioiodinated human von Willebrand factor to Bernard-Soulier, thrombasthenic and von Willebrand's disease platelets. In: Thromb. Res. 19/1980, S. 21–7. PMID 6969462
Weblinks
- Bernard-Soulier-Syndrom bei Online Mendelian Inheritance in Man
- Royal College of Surgeons in Ireland: Bernard-Soulier Syndrome
- Bernard-Soulier Syndrome Organization
- onkodin.de: Hereditäre Thrombozytopenien, Abbildung von Makrothrombozyten bei BSS

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krankheitsbild in Hämatologie und Onkologie
- Erbkrankheit
Wikimedia Foundation.