- Fazioskapulohumerale Muskeldystrophie
-
Klassifikation nach ICD-10 G71.0 Muskeldystrophie ICD-10 online (WHO-Version 2011) 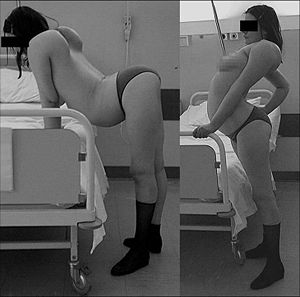 27-jährige Patientin mit FSHD
27-jährige Patientin mit FSHD
Die fazioskapulohumerale Muskeldystrophie (FSHMD, FSHD, Muskeldystrophie Landouzy-Dejerine) ist eine Muskelerkrankung (Myopathie). Der lateinische Name der Erkrankung leitet sich von den hauptsächlich betroffenen Muskelgruppen ab: der Gesichtsmuskulatur (-fazio), der Schultergürtelmuskulatur (-skapulo) und der Oberarmmuskulatur (-humeral). Die Erkrankung wird durch eine Veränderung des Erbguts (Genom) verursacht und wird autosomal dominant vererbt. In den meisten Fällen beginnt die Erkrankung im jugendlichen oder im jungen Erwachsenalter. Der Verlauf ist im Vergleich zu vielen anderen Muskeldystrophien in den meisten Fällen relativ milde. Allerdings benötigen etwa 20 Prozent der Patienten im Verlauf einen Rollstuhl zur Bewältigung längerer Gehstrecken. Der Verdacht auf eine FSHD kann in den meisten Fällen bereits aufgrund des typischen Verteilungsmusters in Verbindung mit dem Beschwerdebild gestellt werden. Gesichert wird die Diagnose mit Hilfe einer molekulargenetischen Untersuchung. Der Schwerpunkt der Behandlung liegt auf regelmäßiger Krankengymnastik. Eine Heilung ist nicht möglich.
Inhaltsverzeichnis
Medizingeschichte
Die Erstbeschreibung der Erkrankung 1868 geht auf Guillaume-Benjamin Duchenne, einem französischen Physiologen.[1] Benannt wurde die Erkrankung allerdings nach den französischen Neurologen Louis Théophile Joseph Landouzy und Joseph Jules Dejerine, die das Krankheitsbild 1884 sehr detailliert beschrieben.[2][3]
Häufigkeit
Die Häufigkeit (Prävalenz) der FSHD wird auf einen Erkrankten pro 20000 Einwohner geschätzt.[2] Sie ist damit nach der progressiven Muskeldystrophie Typ Duchenne und der myotonen Muskeldystrophie Curschmann-Steinert die dritthäufigste erbliche Muskelerkrankung.[4]
Genetische Ursache
Die FSHD ist assoziiert mit einer teilweisen Deletion des polymorphischen Abschnitts D4Z4 auf Chromosom 4, der für das Doppelhomöobox-Protein DUX4 codiert. Die Folge ist die Translation von Spleißvarianten von DUX4, die sich vom normalen DUX4 unterscheiden. Diese Unterschiede machen sich vor allem darin bemerkbar, dass DUX4 nun nicht mehr oder in geringerer Menge in Myoblasten exprimiert wird. DUX4 ist ein Transkriptionsfaktor, der in Myoblasten normalerweise die Expression mehrerer Proteine anregt, die für die Differenzierung der Myoblasten in ausgewachsene Muskelzellen unentbehrlich sind, unter anderem auch die schwere Kette des Myosin. Aus den Muskelvorläuferzellen können also keine Muskeln wachsen.[5]
Krankheitsbild
Sowohl der Beginn der Beschwerdesymptomatik – die Krankheitsmanifestation – als auch Ausprägungsgrad der Beschwerden sind insgesamt sehr variabel. Die Krankheit kann sich beinahe in jedem Lebensalter manifestieren. Typisch ist jedoch ein Beginn in der ersten oder zweiten Lebensdekade. Bei etwa einem Fünftel liegt der Erkrankungsbeginn bereits im Säuglingsalter. Dies ist häufig mit einem schwereren und rascheren Krankheitsverlauf assoziiert. Der Ausprägungsgrad der Symptomatik reicht von leichten Gesichtslähmungen bis zu schweren Lähmungen fast der gesamten Muskulatur.
Die Erkrankung zeigt sich in der Regel zunächst mit einer Schwäche der Gesichtsmuskulatur (mimische Muskulatur). Die Muskulatur ist, insbesondere zu Beginn der Erkrankung, häufig asymmetrisch betroffen. Typischerweise leiden die Patienten durch Lähmung der Augenringmuskeln an einem verminderten Lidschluss (Lagophthalmus), der im Frühstadium dazu führen kann, dass die Patienten mit offenen Augen schlafen. Im Rahmen der körperlichen Untersuchung ist der inkomplette Lidschluss zunächst als Zilienzeichen (Signe de cils) nachweisbar, das heißt, dass bei festem Zukneifen der Augen im Gegensatz zum Gesunden die Wimpern sichtbar bleiben. Ein weiteres Merkmal ist, dass die betroffenen Patienten nicht pfeifen und nicht durch einen Strohhalm trinken können (durch Lähmung des Musculus orbicularis oris). Eine häufig anzutreffende Hypertrophie des Musculus orbicularis oris führt zu einem sogenannten Scapula alata bezeichnet. Ein weiteres Kennzeichen ist eine Horizontal- oder seitlich abfallende Stellung der Schlüsselbeine. Auch eine Atrophie der Brustmuskulatur (Musculi pectorales majores und Musculi pectorales minores) ist typisch, so dass es zu einem Abflachen der Brust kommt.
Anders, als es der Name der Erkrankung suggeriert, ist relativ frühzeitig meist auch die Fußhebermuskulatur betroffen. Aufgrund der Fußheberschwäche zeigen die betroffenen Patienten ein typisches Gangbild, das als Steppergang bezeichnet wird. Erst relativ spät ist die Oberarmmuskulatur betroffen. Darüber hinaus kann, meist in späteren Krankheitsstadien, auch die Beckengürtelmuskulatur betroffen sein.
In fortgeschrittenen Stadien haben die Patienten Schwierigkeiten beim Treppensteigen, beim Kopfüberarbeiten und Probleme beim Aufrichten aus dem liegen. Bei durchschschnittlich 20 Prozent aller Patienten ist im Verlauf ein Rollstuhl zur Fortbewegung notwendig.
Neben der Skelettmuskulatur tritt bei vielen Patienten eine Hörminderung auf, die in den meisten Fällen jedoch nur sehr gering ausgeprägt ist und von den Patienten daher oft gar nicht bemerkt wird. Bei Manifestation der FSHD im Säuglings- oder im Kleinkindalter kann der Hörverlust vereinzelt bis zur Taubheit reichen. Darüber hinaus ist häufig eine Anomalie der Netzhautgefäße nachweisbar, die sich jedoch klinisch selten bemerkbar macht. In seltenen Fällen tritt jedoch ein sogenanntes Coats-Syndrom auf, eine Schädigung der Netzhaut (Retinopathie) mit Teleangiektasien, die zur Blindheit führen kann. Ebenfalls selten und eher untypisch im Vergleich zu anderen Muskelerkrankungen ist eine Beteiligung der Herzmuskulatur. In Einzelfällen kann es zu Herzrhythmusstörungen wie Vorhofflattern und Vorhofflimmern und Reizleitungsstörungen wie Störungen des Sinusknoten kommen.
Literatur
- F. Jerusalem, S. Zierz: Muskelerkrankungen. Thieme-Verlag, 3. Auflage 2003, S. 103-106 ff. ISBN 978-3-13-567803-0
- J. Sieb, B. Schrank: Neuromuskuläre Erkrankungen. Kohlhammer Verlag, 1. Auflage 2009, S. 93-104. ISBN 978-3-17-018381-0
Einzelnachweise
- ↑ Duchenne GBA: Recherches sur la paralysie musculaire pseudo-hypertrophique, ou paraly- sie myo-sclérosique. Arch Gén Méd, 6 ser. 1868;6 ser,11:5–25, 179-209, 305-321, 421-443, 552-588
- ↑ a b Krasnianski M et al.: Facioscapulohumeral muscular dystrophy. The spectrum of clinical manifestations and molecular genetic changes. Nervenarzt. 2003 Feb;74(2):151-8. PMID 12596016
- ↑ L. Landoyzy, J. Dejerine: De la myopathie atrophique progressive (myopathie héréditaire, débutant dans l’enfance par la face, sans altération du système nerveux). Comptes rendus de l’Académie des sciences, Paris, 1884, 98: 53-55.
- ↑ Lunt PW et al.: Genetic counselling in facioscapulohumeral muscular dystrophy. J Med Genet. 1991 Oct;28(10):655-64. PMID 1941962
- ↑ DUX4 bei Online Mendelian Inheritance in Man
Weblinks
Fazioskapulohumerale Muskeldystrophie bei Online Mendelian Inheritance in Man
Wikimedia Foundation.