- Chediak-Higashi-Syndrom
-
Das Chediak-Higashi-Syndrom (CHS) ist eine sehr seltene Erbkrankheit, die vermehrt in jüdischen Bevölkerungsgruppen beobachtet wird.[1]
Inhaltsverzeichnis
Diagnose
Das Chediak-Higashi-Syndrom manifestiert sich mit okulo-kutanem Albinismus (verminderte Pigmentierung), silbrig-blondem Haar, Hepatosplenomegalie, Ganglion-Hypertrophie und rezidivierenden eitrigen Infektionen der Haut und der Atemwege.
Im peripheren Blutbild ist eine Granulationsanomalie der Leukozyten und Lymphozyten (Riesengranula), sowie plasmatische Einschlusskörperchen in den myeloischen Zellen im Knochenmark sichtbar. In der pränatalen Diagnostik kann die Krankheit durch eine fetale Blutuntersuchung oder eine Haut-Haar-Biopsie festgestellt werden.
Ursache
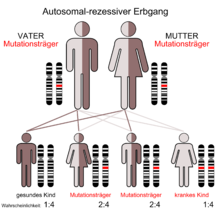 Der autosomal-rezessive Erbgang
Der autosomal-rezessive Erbgang
Das Syndrom wird autosomal-rezessiv vererbt. Das CHS-Gen liegt auf dem langen Arm von Chromosom 1, Genlocus q42.1–q42.2. Ursachen der Symptome sind Funktionsstörungen der polymorphkernigen Leukozyten (sie enthalten charakteristische große lysosomale Vakuolen) und ein Mangel an Natural-Killer (NK)-Lymphozyten. Die Untersuchung eines Tiermodells, der beige-Maus, führte zur Aufdeckung der Ursache, Mutationen im Gen für das LYST-Protein, dessen Funktion noch nicht bekannt ist.
Mit dem Chediak-Higashi-Syndrom ist meist eine aggressiv verlaufende Parodontitis verbunden.[1]
Therapie
Die Erkrankung kann mittels Knochenmarkstransplantation therapiert werden.[1]
Prognose
Die Lebenserwartung ist stark verringert. Die meisten an CHS leidenden Kinder erreichen infolge der Infektionen oder wegen der Ausbildung maligner Tumore nicht das zweite Lebensjahrzehnt.[1]
Siehe auch
Einzelnachweise
- ↑ a b c d B. Kugel u. a.: Parodontitis als Symptom von Syndromerkrankungen. In: Zahnärztliche Mitteilungen, 3/2003, S.46.
Literatur
- J. Wolf u. a.: Das Chediak-Higashi-Syndrom. In: Der Nervenarzt 77/2006, S.148–57.
Wikimedia Foundation.