- Albinotisch
-
Klassifikation nach ICD-10 E70.3 Albinismus ICD-10 online (WHO-Version 2006)  Albino-Pinguin
Albino-Pinguin Tierpräparate im Carl-Schweizer-Museum in Murrhardt
Tierpräparate im Carl-Schweizer-Museum in Murrhardt Albino-Känguru
Albino-KänguruAlbinismus (von lateinisch albus[1] ‚weiß‘) ist eine Sammelbezeichnung für angeborene Störungen in der Biosynthese der Melanine (das sind Pigmente oder Farbstoffe) und der daraus resultierenden helleren Haut-, Haar- und Augenfarbe. Betroffene Tiere nennt man Albinos, betroffene Menschen ziehen meist die neutralere Form „Menschen mit Albinismus“ vor. Menschen mit Albinismus bekommen leichter Sonnenbrand und bekommen deshalb auch leichter Hautkrebs. Außerdem sehen sie unschärfer als Menschen mit normaler Melaninproduktion und das räumliche Sehen funktioniert schlechter.
Albinismus folgt meist einem rezessiven Erbgang und kommt beim Menschen weltweit mit einer Häufigkeit (Inzidenz) von 1:20.000 vor. Häufungen finden sich vor allem in Afrika mit einer Häufigkeit von 1:10.000 und höher.
Offensichtliche Unterschiede
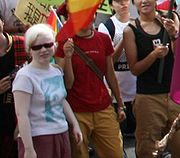 Taiwanesin mit Albinismus
Taiwanesin mit Albinismus Mädchen mit Albinismus in Papua-Neuguinea
Mädchen mit Albinismus in Papua-NeuguineaMenschen mit Albinismus sind an ihrem Äußeren nicht eindeutig als solche zu erkennen. Sie sehen zwar heller aus als Familienmitglieder ohne Albinismus, doch meist ist noch eine Restfunktion der Melaninproduktion erhalten, so dass es auch Schwarze mit Albinismus gibt, die deutlich braune Haut und hellbraune Augen haben. Selbst Menschen, deren Körper überhaupt kein Melanin produzieren kann, die also vollständig albinotisch sind, fallen in Mittel- und Nordeuropa nicht extrem auf, da rosa Haut, weißblondes Haar und blaue oder graue Augen hier auch bei nicht albinotisch veranlagten Menschen vorkommen.
Unterschiede am Sehsystem
Bei Menschen mit Albinismus fehlt im Auge der Farbstoff Melanin (Unterpigmentierung). Das hat mehrere Auswirkungen.
Aufhellung der Augenfarbe bei Albinismus
Die Augenfarbe des Menschen variiert von dunkelbraun über hellbraun und grün bis hin zu blau. Albinismus hellt die Augenfarbe auf. Vollständiger Albinismus führt unabhängig davon, welche Augenfarbe der Betreffende ohne seinen Albinismus hätte, zu roten Augen. Das ist aber beim Menschen sehr selten. Wenn sehr wenig Melanin produziert wird, sind die Augen blau. Schwächer ausgeprägter Albinismus, bei dem der Körper noch merkliche Mengen an Melanin erzeugen kann, hellen die Augen entsprechend weniger auf. So können braune Augen zu hellen braunen, grünen oder blauen Augen und grüne zu blauen aufgehellt werden.
Unscharfes Sehen
Wenn der Körper nahezu kein Melanin produzieren kann und dadurch fast kein Melanin im Auge vorhanden ist, ist die Iris nicht völlig undurchsichtig und das Auge ist besonders lichtempfindlich. Auch die Aderhaut des Auges enthält normalerweise Melanin. Deshalb ist eine ausgeprägte Blendungsempfindlichkeit (Photophobie) typisch für Menschen mit stark ausgeprägtem Albinismus. Wenn man in das Auge hineinleuchtet, zeigt sich diese mangelnde Undurchsichtigkeit durch rote Lichtreflexe. Da Licht nicht nur durch die Pupille hereinkommt, sondern auch die Iris durchdringt, sind die Kontraste zwischen hellen und dunklen Stellen des Raumes nicht so deutlich. Das Farbempfinden ist normal, da Albinismus keinen Einfluss auf die Bildung des Rhodopsins hat.
Auch die Fovea centralis, der Fleck des schärfsten Sehens, ist nicht normal ausgeprägt, da seine Entwicklung ebenfalls durch Melanin beeinflusst wird. Außerdem sind Menschen mit Albinismus oft unfähig, das Auge korrekt scharf einzustellen, viele sind kurz- oder weitsichtig.
Gestörtes räumliches Sehen
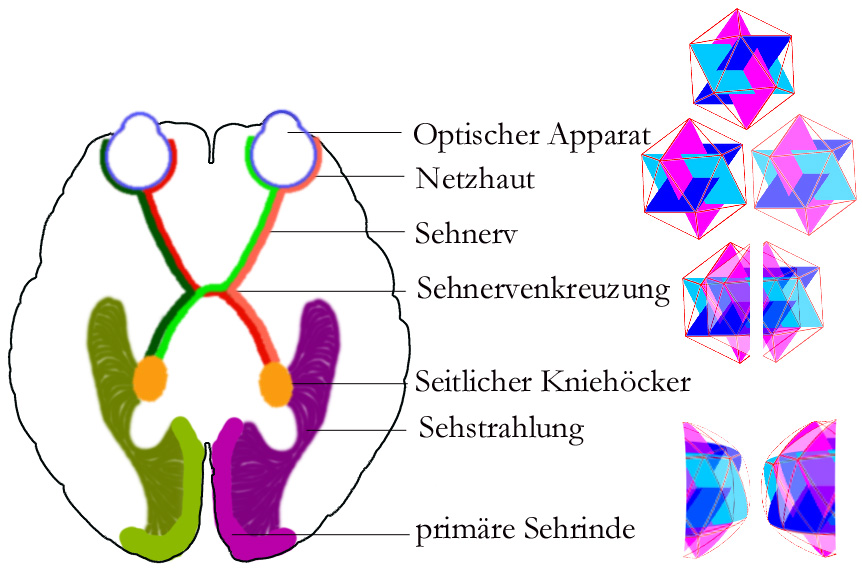
 Sehsystem des Menschen. Zur Veranschaulichung der Verarbeitungswege ist ein Ikosaeder nach den einzelnen Verarbeitungsphasen schematisch dargestellt.
Sehsystem des Menschen. Zur Veranschaulichung der Verarbeitungswege ist ein Ikosaeder nach den einzelnen Verarbeitungsphasen schematisch dargestellt.Außerdem spielt Melanin auch bei der Steuerung der Entwicklung der Sehnerven eine Rolle: Normalerweise ist das Gesichtsfeld beim Menschen unter beiden Gehirnhälften gleichmäßig aufgeteilt – jede Gehirnhälfte hat ihre Seite und bekommt von beiden Augen den Teil des Bildes geliefert, der zu dieser Seite gehört. Durch den Vergleich beider Bilder kann jede Gehirnhälfte die Entfernung der Gegenstände berechnen. Bei Menschen mit Albinismus kreuzt ein größerer Anteil der Sehnerven zur gegenüberliegenden Gehirnhälfte, so dass die zusammengehörigen Bilder nicht immer auf derselben Seite verarbeitet werden. Deshalb ist auch das räumliche Sehen gestört.
Nystagmus
Außerdem tritt bei Menschen mit Albinismus ein Nystagmus (Augenzittern) auf, das selbst wohl einen störenden Einfluss auf das Sehvermögen hat und dessen Ursache noch nicht geklärt ist.
Sonstige Unterschiede
Menschen mit Albinismus bekommen leichter Sonnenbrand und haben deshalb ein höheres Hautkrebsrisiko. Da lichtempfindliche Menschen der Sonne ausweichen oder sich mit Kleidung, Sonnencreme und Sonnenbrille davor schützen, sind Fälle von Hautkrebs bei Albinismus in der Fachliteratur die Ausnahme.
Während die meisten Menschen mit Albinismus eine hellere Augen- und Haarfarbe haben als ihre nicht albinotischen Blutsverwandten (okulokutaner Albinismus, OCA),[2] gibt es auch Fälle von Albinismus, bei denen sich die Symptomatik allein auf die Augenschäden beschränkt, während sie äußerlich normal aussehen (okulärer Albinismus, OA).
Bei nicht albinotischen Weißen ist die helle Hautfarbe auf eine veränderte Regulation der Melaninproduktion zurückzuführen, nicht auf eine Unfähigkeit zur Melaninproduktion. Deshalb sind ihre Augen normal gebaut und enthalten normal viel Melanin, selbst wenn die Iris blau aussieht wie bei Albinos.
Behandlung
Albinismus hat keinen Einfluss auf die geistige Entwicklung von Menschen. Deshalb können sie meist, obwohl der Stoffwechseldefekt nicht therapiert werden kann, mit Hilfe von Sehhilfen, getönten Kontaktlinsen oder entsprechendem Hautschutz ein weitgehend normales Leben führen.
Die Sehbehinderung schwankt auch innerhalb des gleichen Typs stark. Bei manchen Menschen mit Albinismus ist die Sehfähigkeit kaum eingeschränkt, bei manchen jedoch auf ca. 10 % im Vergleich zur Normalsichtigkeit. Damit kann man, so lange die Verkehrssituation übersichtlich ist, Fahrradfahren, übersieht aber oft selbst so große Dinge wie die Stangen am Eingang eines Fußgängerweges, die Autos den Zugang verwehren sollen. Autofahren ist deshalb nur in wenigen Ländern mit vielen Einschränkungen erlaubt. Gesichter zu erkennen ist aus einer Entfernung von mehreren Metern unmöglich, dafür werden oft die Gangart oder prägnante Kleidungsstücke erkannt. Zum Lesen muss der Text meist deutlich vergrößert werden.
Physiologie des Albinismus
Melaninsynthese
Der Farbstoff Melanin wird von farbstoffbildenden Zellen, den Melanozyten produziert. Die Vorstufen der Melanozyten, die Melanoblasten, wandern während der Schwangerschaft in der frühen Fötalperiode aus der Neuralleiste in die Epidermis der Haut, in die Haarfollikel und verschiedene andere Organe aus. In der Haut angelangt, differenzieren sich die Melanoblasten zu Melanozyten und bilden zahlreiche Zellfortsätze aus, über die sie das Melanin an die Keratinozyten weitergeben. Die Menge der Melanozyten ist bei Schwarzen dieselbe wie bei Weißen und auch ein an Albinismus Erkrankter hat normal viele Melanozyten. Die Hautfarbe wird durch die Menge und Qualität des gebildeten Farbstoffs Melanin bestimmt, nicht durch die Anzahl dieser Zellen.
Melanozyten enthalten Melanosomen, kleine membranumschlossene Bläschen, in denen der Farbstoff Melanin produziert wird. Sie sind in ihrer Funktion den Lysosomen (Zellorganellen, die der Verdauung dienen) sehr ähnlich, denn beide enthalten Stoffe, die für die Zelle gefährlich sind und deshalb nicht mit dem Rest der Zelle in Berührung kommen dürfen. Die Lysosomen beinhalten eiweißauflösende Enzyme und die Melanosomen Zwischenprodukte der Melaninsynthese wie Chinone und Phenole, die Membranen der Zelle beschädigen können.
Um Melanin zu produzieren, werden diverse Enzyme gebraucht, die nacheinander beim Aufbau des Melanins mitwirken. Wenn eines der Enzyme dieser Genwirkkette nicht mehr funktionsfähig ist, tritt Albinismus auf. Die Eumelaninbildung in den Melanosomen beginnt mit einer Hydroxylierung der Aminosäure (AS) L-Tyrosin durch das membranständige Enzym Tyrosinase. Neben diesem Schlüsselenzym sind zwei weitere ebenfalls membranständige Enzyme DHICA-Oxidase und Dct nötig, damit Eumelanin gebildet werden kann.
Molekulargenetische Klassifizierung des Albinismus
Obwohl Unterschiede im Aussehen der Menschen mit Albinismus schon früh beschrieben wurden, ging man davon aus, dass Albinismus auf die Veränderungen in einem einzigen Gen zurückzuführen ist. Erst die von Trevor-Roper im Jahre 1952 beschriebene Familie, bei der beide Eltern von Albinismus betroffen waren und dennoch normal pigmentierte Kinder hatten, gab einen ersten Hinweis auf die genetische Heterogenität dieser Erkrankung. Beide Eltern waren in diesem Fall homozygot für Genmutationen, die zum Albinismus führten. Diese betrafen jedoch unterschiedliche Gene, so dass die Kinder für jede der beiden Mutationen heterozygot waren und somit klinisch nicht von Albinismus betroffen waren.
Zuerst klassifizierte man Albinismus nach dem äußeren Erscheinungsbild. Später konnte man nachweisen ob Tyrosinase – ein zur Melaninproduktion nötiges Enzym – vorhanden war. Mit der Möglichkeit der Identifizierung einiger verantwortlicher Gene für den OCA etablierte sich schließlich eine molekulargenetische Klassifikation. Man stellte fest, dass die unterschiedlichen Phänotypen nicht immer auf Mutationen in unterschiedlichen Genen zurückzuführen sind, sondern oft unterschiedliche Ausprägungen diverser Mutationen in einem Gen darstellen. Die klinische Differenzierung bleibt schwierig, da nicht vom Aussehen (Phänotyp) auf die verursachende Mutation (Genotyp) geschlossen werden kann.
Okulokutaner Albinismus Typ 1
Okulokutaner Albinismus Typ 1, abgekürzt OCA 1, oculo-cutan setzt sich zusammen aus lat.: oculus=Auge und cutaneus=die Haut betreffend. Der Begriff dient zur Unterscheidung vom Okulären Albinismus, bei dem nur die Augen sichtlich verändert sind. Andere Bezeichnungen hierfür sind Albinismus totalis 1, Tyrosinase-Gen-bezogener Albinismus.
Der okulokutane Albinismus Typ 1 wird durch Mutationen im Tyrosinase-Gen verursacht. Da 40 % aller von OCA Betroffenen OCA 1 haben, ist es die zweithäufigste Albinismusform. Das Tyrosinase-Gen befindet sich auf Chromosom 11 (11q14-21), beinhaltet fünf Exons und codiert für das Protein Tyrosinase mit einer Länge von 529 Aminosäuren.
Tyrosinase hat eine Schlüsselfunktion bei der Melaninsynthese. Je nachdem ob die Tyrosinase durch die Mutation völlig funktionsunfähig wird oder teilweise noch seine Funktion in der Melaninsynthese erfüllt, unterscheidet man zwei Formen von OCA1:
Menschen mit Tyrosinasenegativer OCA 1 (OCA 1A) haben völlig funktionsunfähige Tyrosinase und deshalb kein Melanin. Betroffene kommen mit weißen Haaren, heller Haut und blauen, durchscheinenden Iriden zur Welt. Sie entwickeln kein Melanin im Laufe des Lebens. OCA 1A gibt es bei allen Rassen des Menschen und bei zahlreichen Tierarten. Außerdem haben sie meist eine starke Sehbehinderung.
Bei Tyrosinasepositivem OCA 1 (OCA 1B), kann die Tyrosinase Melanin produzieren, aber nicht so gut wie normal. Die Betroffenen kommen wie bei OCA 1A ohne Pigmentierung der Haut, der Haare und der Iris zur Welt. Im Verlauf des Lebens bilden sie aber etwas Melanin. Menschen mit OCA 1B können sehr unterschiedlich aussehen, je nachdem wie gut die Tyrosinase noch funktioniert, auch die Sehbehinderung kann in unterschiedlichem Maße ausgeprägt sein. Der OCA 1B wird auch yellow OCA genannt, da das Nachdunkeln bei Betroffenen oft zu gelber Haarfarbe führt. Dies wird auf die Synthese von Phäomelanin zurückgeführt. Bei Phäomelanin handelt es sich im Gegensatz zum schwarzen Eumelanin um einen braunen Farbstoff.
Auch im Tyrosinase-Gen kommen Mutationen vor, die sich nur in den Augen äußern.
Eine andere Mutation erzeugt eine Tyrosinase, deren Funktionsfähigkeit von der Temperatur abhängt. Deshalb sind sind die Haare am Körper im Gegensatz zum kühleren Kopf farblos. Solche Mutationen kommen auch im Tierreich vor, beispielsweise bei Siamkatzen und Russenkaninchen.
Bisher sind über 100 verschiedene Mutationen im Tyrosinase-Gen entdeckt. Einige dieser Mutationen erzeugen zwar eine funktionierende Tyrosinase, deren Reifung im endoplasmatischem Retikulum gestört ist oder zur Störung des Transports von Tyrosinase in die Melanosomen.
In Europa haben 30-40 % der Menschen mit Albinismus OCA1, damit ist es die häufigste aller OCA-Formen. Die Häufigkeit beträgt 1:39.000 bei Kaukasiern und 1:28.000 im afrikanischen Raum.
Okulokutaner Albinismus Typ 2
Mit 50 % Anteil an OCA ist der OCA 2 die häufigste Albinismusform weltweit und kommt bei Afrikanern und Afroamerikanern in einer Häufigkeit von 1:10.000, bei Hopi-Indianern 1:277 vor. In Europa haben ungefähr 10-20 % der Patienten mit OCA einen OCA 2. Der OCA 2 wird auch als Albinismus totalis 2 bezeichnet und entsteht durch Mutationen im P-Gen, welches nach dem homologen pink-eyedilution Gen der Maus benannt wurde. Das P-Gen befindet sich auf dem langen Arm von Chromosom 15 (15q11-13) und kodiert ein Membranprotein der Melanosomen.
Für die Melaninproduktion braucht Tyrosinase einen niedrigen pH-Wert. Das P-Gen gehört zu einem Protein, das bekannten Ionentransportern ähnlich ist und vermutlich den niedrigen pH-Wert in den Melanosomen aufrechterhält. Bei einem durch ein fehlerhaftes Protein erhöhten pH-Wert kann die Tyrosinase nicht mehr richtig arbeiten.
Die Phäomelanosomen sind von Mutationen im P-Gen weniger betroffen, als die Eumelanosomen, deshalb haben Menschen mit OCA 2 gelbliche, blonde oder sogar braune Haare, die im Verlauf ihres Lebens dunkler werden. Die Haut und Haarfarbe von Menschen aus Rassen mit dunkler Haut ist immer heller als die ihrer nicht albinotischen Verwandten. Menschen aus Ländern mit heller Haut und Haarfarbe wie beispielsweise aus Skandinavien, sehen dagegen oft völlig normal aus. Dann können nur noch die typischen Augenfehler zur Diagnose führen. Ein weiterer Phänotyp von OCA 2 ist auch unter dem Namen brauner OCA bekannt. Dieser wurde bisher nur bei Afrikanern und Afroamerikanern beschrieben. Die Betroffenen haben braune Haare, braune oder blau-grüne Iriden und eine hellbraune Haut, die leicht bräunt. Eine Sehbehinderung, Augenzittern und Strabismus werden ebenfalls gefunden.
Okulokutaner Albinismus Typ 3
OCA 3 beruht auf vorzeitigen STOP-Kodons im TYRP-1-Gen (5,6-Dihydroxyindol-2-carbonsäure-Oxidase). Das TYRP-1-Gen ist auf Chromosom 9p23 lokalisiert. Andere Namen des Gens sind: CAS2, CATB, GP75, TRP, TRP1, TYRP, b-PROTEIN. Es entspricht dem sog. braunen Gen der Maus auf Chromosom 4, da Mutationen in diesem Gen bei der Maus zur braunen Fellfarbe führen. DHICA-Oxidase ist der Tyrosinase sehr ähnlich, findet sich in der Membran der Melanosomen und unterstützt die Tyrosinase in ihrer Arbeit.
Der okulokutane Albinismus Typ 3 (OCA 3) wurde erstmals 1996 beschrieben. Er führt meist zu einer roten OCA (rufous OCA oder ROCA) mit roter oder rotbrauner Haut, ingwerfarbenen oder roten Haaren und einer haselnussfarbenen Iris. Die Veränderungen am optischen Apparat sind oft weniger stark und seltener ausgeprägt als bei den anderen OCA Typen. Im Gegensatz dazu wurde beim zuerst beschriebenen Fall eine braune OCA gefunden. Da eine weitere Untersuchung des Zwillings in seinem späteren Leben nicht erfolgte, kann es sein, dass sich sein Aussehen später zur roten OCA weiterentwickelt haben könnte.
Okulokutaner Albinismus Typ 4
Der okulokutane Albinismus Typ 4 (OCA 4) wurde erst 2001 als eine neue Form des OCA beschrieben. Ausgehend von der Beobachtung, dass es bei der Maus durch Mutationen im underwhite Gen bzw. im Matp-Gen auf Chromosom 15 zur Hypopigmentierung kommt, vermutete man einen Zusammenhang des entsprechenden Gens beim Menschen mit OCA. Das MATP-Gen beim Menschen wurde auf Chromosom 5 gefunden. Weitere Namen des Gens sind: 1A1, AIM1, SLC45A2 und MATP. Möglicherweise sind beide Proteine für die Regulation des pH-Werts in den Melanosomen verantwortlich. Bei deutschen mit Ocolucutanem Albinismus macht dieser Typ etwa 3 % der Fälle aus. In der japanischen Bevölkerung kam sie in 24 % der OCA-Fälle vor. In dieser Untersuchung war OCA 4 sogar häufiger als OCA 2 (8 %) und ist damit nach OCA 1 die zweithäufigste Albinismusform in Japan.
Okulärer Albinismus Typ 1
Der okuläre Albinismus Typ-1 (OA 1) wird X-chromosomal rezessiv vererbt. Die männlichen Betroffenen zeigen klinisch alle typischen Veränderungen des optischen Apparates beim Albinismus. Die weiblichen Überträger sind nur an der stellenweisen Hypopigmentierung ihrer Netzhaut zu erkennen. Alle Melanozyten, sowohl in den Augen als auch in der Haut, weisen Makromelanosomen auf, wodurch man OCA und OA morphologisch unterscheiden kann. Man vermutet, dass das betroffene Protein ein intrazellulärer GProtein-gekoppelter Rezeptor ist und zur Melanosomentwicklung beiträgt.
Unbekannte Mutationen
Bei der Maus sind bisher über 100 Gene bekannt, die die Fell- und Augenfarbe beeinflussen. Daher ist davon auszugehen, dass es auch im menschlichen Genom noch einige bisher unbekannte Gene gibt, die die Pigmentierung beeinflussen.
Syndrome, die mit Albinismus verbunden sind
Während die meisten Menschen mit Albinismus nur eine hellere Haut und eine Sehbehinderung haben, gibt es einige Erbkrankheiten, bei denen der Albinismus mit weiteren Krankheitssymptomen vergesellschaftet auftritt.
OCA 2 assoziierte Syndrome
Zwei mit OCA 2 assoziierte Syndrome sind bekannt: das Prader-Willi-Syndrom (PWS) und das Angelman-Syndrom. Beide beruhen auf Mutation auf dem langen Arm von Chromosom 15, wo auch das P-Gen liegt, das für OCA 2 verantwortlich ist.
Prader-Willi-Syndrom (PWS)
Das Prader-Willi-Syndrom ist eine Entwicklungsstörung, welche durch niedrigen Blutdruck der Neugeborenen, kleine Hände und Füße, Übergewicht, unterentwickelte Hoden oder Eierstöcke und geistige Behinderung gekennzeichnet ist. Die Hälfte der Betroffenen PWS haben eine helle Haut- und Augenfarbe, aber nur einige davon haben die typischen Veränderungen am Auge und den zugehörigen Nerven, haben also OCA 2.
Angelman-Syndrom
Das Angelman-Syndrom ist ebenfalls eine Entwicklungsstörung. Die geistige Behinderung ist schwerer als beim PWS. Sie haben eine verringerte Muskelspannung, eine gestörte Bewegungskoordination, verkleinertes Gehirn, ein charakteristisches unmotiviertes Lachen (Happy Puppet Syndrom) und helle Haut und Haare. Der Anteil von hypopigmentierten Individuen mit Angelman-Syndrom unter allen von Angelman-Syndrom Betroffenen ist nicht bekannt.
Syndrome, die auf Fehlsortierung der Enzyme zurückzuführen sind
Beim Hermansky-Pudlak-Syndrom, dem Griscelli-Syndrom und dem Chédiak-Higashi-Syndrom (CHS) ist das Gen für AP-3 mutiert, ein Eiweiß das eine Funktion beim Transport verschiedener Enzyme in die Lysosomen und Melanosomen hat. Dadurch kommt die Tyrosinase nicht oder nicht in ausreichender Menge in den Melanosomen an, was zu Albinismus führt. Gleichzeitig sind auch andere Funktionen in den Lysosomen mit geschädigt, was weitere Krankheitszeichen verursacht.
Griscelli-Syndrom
Kinder mit Griscelli-Syndrom haben silber-graues Haar und Immundefekte. Wird die Erkrankung nicht behandelt, verläuft sie tödlich. Krankheitsschübe mit Fieber und eindringen von Lymphozyten in Organe führen zu Lebervergrößerung, Erkrankungen der Lymphknoten, starker Verminderung aller Blutzellen und unterschiedlichen sich dauernd verstärkenden Erkrankungen des Nervensystems. Die Krankheitsschübe konnten durch medikamentöse Therapie nur gemildert, nicht aber verhindert werden. Die einzige therapeutische Option ist die Knochenmarktransplantation. Das Griscelli-Syndrom ist selten. Bei Auftreten von grauen Haaren im Kindesalter aber eine rasch abzuklärende Differenzialdiagnose, da es um so besser behandelbar ist, je früher die Krankheit erkannt wird. Eine Variante der Erkrankung wird verursacht durch eine Mutation im Gen für Rab27a.
Hermansky-Pudlak-Syndrom (HPS)
Das Hermansky-Pudlak-Syndrom zeigt klinisch einen OCA, eine Thrombozytenaggregationsstörung mit einer Blutungsneigung und die Akkumulation von Zeroid vor allem in den Zellen der Lunge und des Darms, was zu einer Lungenfibrose und einer granulomatösen Colitis führen kann. Das Syndrom ist weltweit beschrieben worden mit einer ungewöhnlichen Häufung in der Bevölkerung von Puerto Rico, die dort 1:1800 beträgt. Der OCA kann sich klinisch in allen Facetten der OCA-Phänotypen präsentieren. Beim Menschen sind mittlerweile sechs verschiedene HPS-Gene identifiziert worden, deren Genprodukte in den meisten Fällen noch unbekannt sind.
Chédiak-Higashi-Syndrom (CHS)
Das Chediak-Higashi-Syndrom (CHS) ist eine seltene autosomal-rezessiv vererbte lysosomale Speicherkrankheit. Typische Symptome sind wiederkehrende Infekte, ein okulokutaner Albinismus, vermehrte Blutungsneigung und neurologische Ausfälle. Die Pigmentierung von Haut, Haaren und Iriden ist herabgesetzt. Die Betroffenen zeigen oft keinen eindeutigen OCA-Phänotyp, so dass nur durch den Vergleich innerhalb der Familie eine Hypopigmentierung auffällt. Veränderungen des optischen Apparates sind nicht immer nachweisbar.
Gesellschaft
Wie alle Menschen, die andersartig sind, haben Menschen mit Albinismus in dem Maße, wie sie sich von ihren Mitmenschen unterscheiden, ein erhöhtes Risiko, ausgegrenzt und diskriminiert zu werden. Teilweise wurden sie als Kuriositäten im Zirkus vorgeführt oder man sprach ihnen übersinnliche Kräfte zu. Bei hellhäutigen Völkern ist ihre Diskriminierung weniger ausgeprägt, da die äußerlichen Unterschiede geringer, teilweise fast nicht zu erkennen sind.
Bei dunkelhäutigen Völkern sind die Unterschiede augenfälliger, und Ausgrenzung ist deshalb häufiger. Häufig stehen Menschen mit Albinismus in Verruf, Unglück zu bringen (wie etwa im Sudan oder in Mali); siehe zu diesem Thema auch die Biografie des Musikers Salif Keïta. In Tansania ist hingegen in jüngerer Zeit der Aberglaube aufgekommen, dass Albinos glücksbringende Kräfte besäßen. 2007 sollen daher 20 Menschen mit Albinismus von „witch doctors“ getötet worden sein, um aus ihren Körperteilen Zaubermittel herzustellen, die zu Reichtum verhelfen sollen.[3] Von März bis November 2008 waren es sogar 36 Menschen mit Albinismus in Tansania und im benachbarten Burundi, die aus diesem Grund getötet wurden.[4] In Simbabwe diente der Aberglaube, Geschlechtsverkehr mit Albinos würde eine HIV-Infektion heilen, als Vorwand, Frauen mit Albinismus zu vergewaltigen[5].
In Filmen, Büchern und Computerspielen nehmen albinotische Menschen oft die Rolle des Bösen bzw. des Bösewichts ein, wie beispielsweise der mordende Mönch Silas in The Da Vinci Code – Sakrileg. Im Film Powder besitzt der Protagonist zwar besondere Fähigkeiten, die er als „gottgegeben“ annimmt, muss aber trotzdem um soziale Akzeptanz kämpfen.
Albinismus bei Tieren
Bei Tieren sind die genetischen Ursachen und gesundheitlichen Folgen von Albinismus sehr ähnlich gelagert. Bei Tieren wird die Bezeichnung Albino oft ausschließlich für Tiere mit OCA1 verwendet, bei denen keine Restfunktion der Tyrosinase erhalten ist und die deshalb weißes Fell, rosa Haut und rote Augen haben. Das ist verwirrend, da beim Menschen zusätzlich zu allen Mutationen des Tyrosinasegens auch alle anderen Störungen der Melaninsynthese als Albinismus bezeichnet werden, während sie bei Tieren sehr unterschiedliche Namen haben.
OCA1 – der Albino-Locus (C)
 British White Cattle
British White Cattle Auch beim Huhn führt eine Mutation des Albino-Locus zu rezessiv weißer Farbe
Auch beim Huhn führt eine Mutation des Albino-Locus zu rezessiv weißer FarbeDem Ocolucutanen Albinismus Typ 1 beim Menschen entsprechen die verschiedenen Mutationen des C-Locus bei Tieren. Zur Albino-Serie gehören neben völlig weißen Tieren mit roten Augen, die als einzige als Albinos bezeichnet werden, eine Reihe von Allelen, die eine stufenweise Aufhellung von Haut, Haaren und Augen kontrolliert. Ausgehend von einer vollständigen Pigmentierung, wie sie im Wildtyp (C) vorliegt, wird zunächst das Phäomelanin, dann das Eumelanin reduziert.
Während beim Menschen 88 Mutationen des Tyrosinase-Gens beschrieben wurden, ist die Mutation des Allels c der Maus in allen Laborstämmen identisch.
Vollständiger Albinismus ist bei Primaten, Huftieren und Nagetieren weit verbreitet. Auch beim Huhn gibt es ein Tyrosinase-Gen, das mutiert eine rezessive weiße Farbe hervorbringt.
Beim Japan-Reiskärpfling (Oryzias latipes) der zu den Reisfischen zählt wird der Tyrosinaselocus mit i abgekürzt. Es sind mehrere Mutationen dieses Gens bekannt. Es gibt mehrere Albino-Mutanten (i1, i4, and i5) die bei denen ein Transposables Element in das Tyrosinasegen eingefügt ist dadurch Albinismus entsteht, der OCA 1 beim Menschen entspricht. Bei einer weiteren Tyrosinasemutation (i6) entsteht OCA1 dadurch, dass ein Teil des Gens fehlt. Tiere mit der Mutation I1 sind vollständig melanotisch, i4 hat eine geringe Tyrosinaseaktivität und dadurch etwas dunklere Augen. Bei der I5-Mutante ist der Albinismus nur schwach ausgeprägt.[6] [7][8][9]
Bei Nagetieren, Kaninchen, dem Nerz und der Katze gibt es einen als Himalaya oder Colorpoint bezeichneten Farbtyp; bei ihnen funktioniert die Tyrosinase abhängig von der Temperatur, deshalb ist der Körper heller als die kühleren Ohren, Nasenspitze, Schwanz und Pfoten. Ein weiteres Allel führt zur typischen Färbung der Burma-Katze. Auf dieselbe Weise entsteht die Farbe des weißen Gallowayrindes, des British White Cattles und des White Park Cattles.
Der Tyrosinase Locus des Schweins wurde auf Chromosom 9 kartiert. Vollständiger Albinismus ist beim Schwein nicht bekannt. Auf Mutationen des C-Allels werden die schmutzig weiße Farbe des Mangalica-Schweins (Allel ce) und die rezessiv vererbte Aufhellung der Farbe von gelb zu weiß und von rot zu cremefarben beim Berkshire-Schwein (Allel cch) zurückgeführt.
OCA2 – Rosa-Augen Serie (pink eye P)
 Links: Rosa-Augen-Mutation beim Meerschweinchen
Links: Rosa-Augen-Mutation beim Meerschweinchen Astyanax mexicanus
Astyanax mexicanusDem Oculocutanen Albinismus 2 (OCA2) beim Menschen entsprechen die Mutationen der Rosa-Augen-Serie bei Tieren. Mehrere Mutationen dieses Locus führen zu Phänotypen, bei denen das Fell nur aufgehellt ist, während die Augen kein oder fast kein Melanin enthalten und deshalb rot oder rosa erscheinen. Außerdem gibt es Varianten, die Spermienanomalien hervorrufen, aber auch Mutanten mit aufgehelltem Fell und normalfarbenen Flecken. Die Variante pm bildet ein Mosaik zwischen Wildtyp und Aufhellung, ist also gefleckt.
Die Farbaufhellung entsteht dadurch, dass weniger Melanosomen für Eumelanin (Eumelanosomen) gebildet werden, die oft kleiner sind und miteinander verklumpen. In den Melanosomen sind die Farbstoffe nicht so dicht zusammengelagert, so dass die Melanosomen auch nicht ganz so dunkel sind. Von den Melanosomen, die Phäomelanin bilden, gibt es zwar weniger, sie sind aber sonst normal. In der Retina werden keinerlei Pigmente synthetisiert.
In der Schweinerasse Hampshire ist eine Mutation bekannt, die zu roten Augen und einer Aufhellung der schwarzen Farbe zu Grau führte, von dem vermutet wurde, dass es dem P-Locus des Schweins zuzuordnen ist.
Das Allel ps (p-sterile) des P-Locus der Maus führt homozygot zu geringer Körpergröße, Nervosität, Zahnfehlstellungen, frühzeitiger Altersschwäche und Sterilität der männlichen Tiere. Die Sterilität wird aber hauptsächlich durch abnorme Spermien hervorgerufen, geht teilweise aber auch mit schlechter Libido einher. Auch beim Hamster gibt es eine Variante mit Farbaufhellung und Infertilität. Das Allel p (pink-eye dilution) der Maus beeinflusst ebenfalls das Wachstum.
Der Albinismus des Blinden Höhlensalmlers (Astyanax mexicanus) ist auf verschiedene Mutationen zurückzuführen, die zu OCA2 führen.[10]
OCA3 – Braun-Locus (B)
 Mutation des Braunlocus bei einer Katze
Mutation des Braunlocus bei einer Katze Die Braune Farbe der Münsterländer wird durch das Braun-Gen hervorgerufen
Die Braune Farbe der Münsterländer wird durch das Braun-Gen hervorgerufenDer Okulokutane Albinismus vom Typ 3 (OCA3) entspricht dem Braunlocus bei Tieren.
Normalerweise (Allel B) wird das schwarze Eumelanin dicht und regelmäßig in den Melanosomen angeordnet, so dass sie schwarz und eiförmig sind. Durch das rezessive Allel b wird das Melanin aufgelockert, wodurch die Melanosomen braun wirken. Die verschiedenen Mutationen des Braun-Locus hellen deshalb schwarzes Fell zu dunkel- bis hellbraun auf.
Die chemische Erklärung hierzu ist: Bei der Maus oxidiert TYRP1 Dihydroxyindolcarboxylsäure (DHICA) und fördert die Polymerisierung von DHICA Monomeren zu Melanin. Außerdem stabilisiert die DHICA Oxidase die Tyrosinase und andere Melanosomenenzyme.[11]
Auf dem Braun-Locus sind beim Schwein keine Allele bekannt mit der Ausnahme des Allels Bk, das zur Ausprägung brauner Flecke auf rotem Hintergrund führt.
Beim Dexter-Rind wird die hellbraune Farbe durch eine Mutation des TRP1 Gens hervorgerufen.
Braune Mäuse, Ratten und Kaninchen sind signifikant schwerer als schwarze.
Bei vielen Jagdhunderassen gibt es Tiere die durch eines oder mehrere der Allele des Brown-Locus von schwarz zu Braun oder rot aufgehellt wurden. Es gibt vier Allele bs, bd bc und der dominante, nicht aufgehellte Wildtyp B.
OCA4 – MATP-Gen
 Palomino, Quarter Horse
Palomino, Quarter HorseBeim Pferd ist kein vollständiger Albinismus bekannt. Das Creme-Gen (Cr) führt zu unvollständigem Albinismus (OCA4), der unter anderem für die Pferdefarben Buckskin, Palomino, Cremello und Perlino verantwortlich ist. Das Cream-Gen wird teilweise als Dilute-Gen geführt.
Bei der Maus ist MATP für die Mutationen Underwhite und Dominant Brown verantwortlich.
Bei Hühnern (hier SLC45A2 genannt) ruft Mutationen des Gens, das dort auf dem X-Chromosom liegt, die Farben Silver und "Sex linked Imperfekt Albinism" hervor.
Weitere Gene
Auch bei Tieren gibt es weitere Gene, die zu teilweisem oder vollständigen Albinismus führen.
 Links: unaufgehellte Farbe, rechts: durch Champagne aufgehellt
Links: unaufgehellte Farbe, rechts: durch Champagne aufgehelltDas Gen für die Fellfarbe Champagne des Pferdes ist eine Mutation des SLC36A1-Gens (Solute Carrier 36 family A1), das auch PAT1 (proton/amino acid transporter 1) oder LYAAT1 (lysosomal amino acid transporter 1) genannt wird. Es gehört zu derselben Genfamilie wie MATP, das auch SLC45A2 genannt wird.
Eine Mutation des Gens SLC45A5 ist verantwortlich für die "Golden (gol) dilution" genannte Mutation der Maus.
Syndrome, die auf Fehlsortierung der Enzyme zurückzuführen sind
Ähnliche Krankheiten wie das Chédiak-Higashi-Syndrom (CHS), das Hermansky-Pudlak-Syndrom (HPS) gibt es bei der Maus, beim Aleutennerz, der Perserkatze, beim Rind, beim Schwein, bei der Ratte, beim Fuchs und selbst beim Killerwal.
Auch die zyklische Hämatopoese des Hundes ähnelt diesen Syndromen. Betroffene Tiere sind silbergrau, die der neutrophilen Granulocyten sind periodisch verringt, eben so die roten Blutkörperchen (Erythrozytopenie) und die Blutplättchen (Thrombozytopenie). Das führt zu einer Blutgerinnungsstörung und erhöhter Infektanfälligkeit. Meist sterben die Tiere kurz nach der Geburt.
Griscelli-Syndrom – Aufhellung (dilution D)
Neben den dem Griscelli-Syndrom entsprechenden Krankheiten werden vom Dilution-Lokus (D) bei Tieren auch diverse Farbaufhellungen hervorgerufen, die nicht mit ernsthaften Erkrankungen verbunden sind.
Weitere Mutationen, die mit Albinismus verbunden sind
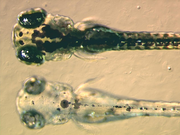 Larven des Zebrabärblings, oben wildfarben, unten Mutante "bleached blond"
Larven des Zebrabärblings, oben wildfarben, unten Mutante "bleached blond"Die Mutante "bleached blond" des Zebrabärblings haben eine Mutation in einem Gen das den Teil Ac45 der ATP-Synthase herstellt. Als Embryos erscheinen sie, abgesehen von ihrer aufgehellten Farbe, völlig normal. Die meisten der Larven mit dieser Mutation entwickeln aber während des weiteren Wachstums keine Schwimmblase und gedeihen nicht so recht, auch wenn sie einige Tage überleben können.[12]
Soziale Folgen von Albinismus für Tiere
Auch viele Tiere grenzen abweichend aussehende oder sich ungewöhnlich verhaltende Artgenossen so aus, wie wir es von Menschen kennen.
Ganz anders ist die Situation in Menschenobhut: Da die Tiere von ihren Besitzern beschützt und mit Futter versorgt werden, ist die weiße Farbe für das Überleben unerheblich. Auch schlechtere Augen dürften nur für Pferde ein ernsthafter Nachteil sein, da diese ja oft mit Reiter galoppieren und springen müssen und sich dadurch die Verletzungsgefahr erhöht. Die mit Albinismus oder Leuzismus verbundene größere Zahmheit der Tiere bietet dagegen einen erheblichen Überlebensvorteil: sie macht es Menschen leichter eine Beziehung zu den Tieren aufzubauen. Die weit verbreitete Ansicht, dass helle Farbschläge mit verminderter Aggressivität einhergehen, wird durch den gemeinsamen Syntheseweg von Adrenalin und Dopachinon aus Dopa untermauert. Albino-Mäuse bringen ihre Jungtiere häufiger und zuverlässiger in das Nest zurück.
Tiere mit Albinismus sind oft Publikumslieblinge in Zoos. Albino-Tiere sind auch äußerst beliebt für Tierversuche, denn die pigmentlose Haut eignet sich angeblich besser dafür.
Selektion
Die fehlende Tarnung, das durch Albinismus eingeschränkte Sehvermögen und die Lichtempfindlichkeit sind Selektionsnachteile. Bei Mäusen wurde eine deutlich verminderte Laufleistung und weniger Aktivität in offenem Gelände festgestellt.
Bei Tieren die ihr gesamtes Leben in Höhlen verbringen wie der Blinde Höhlensalmler (Astyanax mexicanus) hat die Tarnfarbe keine Funktion mehr, da dort kein Licht ist, in dem man die Farbe wahrnehmen könnte. Inzwischen sind von über 80 Fischarten albinotische Höhlenformen bekannt.[13] [14]
Ursachen von ähnlichen Symptomen ohne Vorhandensein von Albinismus
- Ebenfalls zu Störungen der Melaninsynthese führen Mutationen des Silver-Locus.
- Für Leuzismus sind unterschiedliche Gene verantwortlich, im frühen Embryonalstadium zu einer mangelhaften Wanderung der Zellen aus der Neuralleiste in den Körper führen.
- Wie bei Kühen bekannt, können die meisten Tiere und auch Menschen gescheckt sein. Diese Eigenschaft entsteht meist durch Leuzismus und ist ebenfalls gelegentlich mit Taubheit und Augenfehlern verbunden.
- Vitiligo oder die Weißfleckenkrankheit äußert sich durch weiße, pigmentfreie Hautflecken, die durch Absterben der Melanozyten langsam größer werden.
- Die Tuberöse Hirnsklerose oder das Bourneville-Pringle-Syndrom ist eine autosomal-dominant vererbte Krankheit mit einer Häufigkeit von 1:20 000–1:40 000 in der Bevölkerung. Sie zeigt sich durch Adenoma sebaceum (viele kleine knötchenförmige Tumore auf der Gesichtshaut und unter den Fingernägeln), Epilepsie, zunehmende geistige Behinderung und weiße Flecken auf der Haut. Diese Flecken sind darauf zurückzuführen, dass in den Melanozyten die Melanosomen zwar angelegt werden aber nicht vollständig ausreifen und deshalb hell bleiben.
- Phenylketonurie ist eine erbliche Stoffwechselstörung, die unbehandelt zu schwerer geistiger Retardierung und auch zu heller Haut-, Haar- und Augenfarbe führt.
Sonstige Farbabweichungen
- Bei Pflanzen führt der Mangel am grünen Farbstoff Chlorophyll zu Panaschierung.
- Schwärzlinge mit Überproduktion von Melanin: Melanismus
- Rothaarige: Es gibt zwei Arten von Melanin. In den Eumelanosomen wird das braunschwarze Eumelanin hergestellt, in den Phäomelanosomen das gelbrote Phäomelanin. Rothaarige produzieren hauptsächlich Phäomelanin während Menschen mit braunen bzw. schwarzen Haaren mehr Eumelanin herstellen. Eumelanin dient dem Schutz vor UV-Strahlung, während unbekannt ist, ob Phäomelanin eine nützliche Funktion hat.
- Fellfarbe
Tiere mit Albinismus
- Copito de Nieve (Gorilla)
Literatur
Albinismus bei Menschen
- Aleksandra Lipka: Albinismus: Mutationssuche im TRP-1-Gen. Universität zu Lübeck 2004 (Dissertation)
- P. M. Lund: Oculocutaneous albinism in southern Africa: Population structure, health and genetic care. In: Annals of Human Biology. Volume 32, Number 2, März/April 2005, S. 168-173
- Barbara Käsmann-Kellner, Thorsten Schäfer, Christof M. Krick, Klaus W. Ruprecht, Wolfgang Reith, Bernd Ludwig Schmitz: Anatomische Unterschiede der Nervi optici, des Chiasmas und der Tractus optici bei normal- und hypopigmentierten Personen: eine standardisierte MRI- und fMRI-Untersuchung. In: Klinisches Monatsblatt Augenheilkunde 220, 2003 S. 334-344
- Charlotte Jaeger, Barrie Jay: X-linked ocular albinism. In: Human Genetics. Volume 56, Number 3, S. 299-304, Februar 1981.
- Birgit Lorenz, Markus Preising, Ulf Kretschmann: Molekulare und klinische Ophthalmogenetik. In: Deutsches Ärzteblatt. 98, Ausgabe 51-52 vom 24. Dezember 2001, Seite A-3445, B-2902, C-2698
- TYRP1 tyrosinase-related protein 1 [ Homo sapiens ]. NCBI, GeneID: 7306, updated 07-Aug-2007, http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=7306&ordinalpos=1&itool=EntrezSystem2.PEntrez.Gene.Gene_ResultsPanel.Gene_RVDocSum
- Markus Kaufmann: Albinismus: Das Tyrosinase-Gen in 78 Variationen Inauguraldissertation zur Erlangung der Doktorwürde der Universität zu Lübeck , im September 2004
- Regine Witkowski, Otto Prokop, Eva Ullrich, G Thiel: Lexikon der Syndrome und Fehlbildungen: Ursachen, Genetik, Risiken. Veröffentlicht von Springer, 2003. ISBN 9783540443056 S. 86-88
- Esdras Ndikumana: Mörderbanden machen Jagd auf Albinos, Spiegel Online, 25. Oktober 2008.
Syndrome aus dem Formenkreis des Albinismus
- J. Wolf, C. Jacobi, H. Breer, A. Grau: Das Chediak-Higashi-Syndrom. In: Der Nervenarzt. Band 77, Nummer 2, Februar 2006, S. 148-157
- Heinrich Burkhardt, Bettina Jung, Steffen J. Diehl, Walter Back, Rainer Gladisch: Ungewöhnliche Ursache einer Lungenfibrose bei einer 71-jährigen Patientin. In: Medizinische Klinik. Ausgabe 97, Nummer 3, März 2002, S. 165-169
- P. Habermehl, S. Althoff, M. Knuf, J.-H. Höpner: Griscelli-Syndrom: ein Fallbericht. I: Klinische Pädiatrie. 215, 2003, S. 82-85
- Björn Chapuy: Analyse der putativen AP-3-Funktion für die Vesikelbildung am Trans-Golgi-Netzwerk. Georg-August-Universität Göttingen, 2005 (Dissertation)
- W. Tilgen: Zur Ultrastruktur der sogenannten White leaf-shaped macules bei der tuberösen Hirnsklerose Bourneville-Pringle. In: Archives of Dermatological Research. Band 248, Nummer 1. März 1973, ISSN 0340-3696, S. 13-27
Albinismus bei Tieren
- Krista Siebel: Analyse genetischer Varianten von Loci für die Fellfarbe und ihre Beziehungen zum Farbphänotyp und zu quantitativen Leistungsmerkmalen beim Schwein. Institut für Nutztierwissenschaften der Humboldt-Universität zu Berlin, Juli 2001 (Dissertation)
- Petra Keller: Untersuchungen zur Entwicklung der frühen akustisch evozierten Potentiale (FAEP) bei der Katze für den Einsatz in der Grundlagenforschung und zur klinischen Anwendung. Tierärztliche Hochschule Hannover, 1997 (Dissertation)
- NCBI: Slc45a2 solute carrier family 45, member 2 (Mus musculus). GeneID: 22293. Stand: 6. April 2007
- Denis Mariat, Sead Taourit, Gérard Guérin: A mutation in the MATP gene causes the cream coat colour in the horse. In: Genetics Selection Evolution. 35, 2003 doi:10.1051/gse:2002039, S. 119–133 119
- Sheila Schmutz: Genetics of Coat Color Patterns in Cattle. Stand: 19. Januar 2005
- Gunnarsson U, Hellstrom AR, Tixier-Boichard M, Minvielle F, Bed'hom B, Ito S, Jensen P, Rattink A, Vereijken A, Andersson L.: Mutations in SLC45A2 cause plumage color variation in chicken and Japanese quail. Genetics. 2007 Feb;175(2):867-77. Epub 2006 Dec 6. PMID: 17151254
- Sheila Schmutz: Dog Coat Color Genetics. Brown. last updated on July 1, 2006
- Chung-Ming Chang, Jean-Luc Coville, Gérard Coquerelle, David Gourichon, Ahmad Oulmouden, and Michèle Tixier-Boichard: Complete association between a retroviral insertion in the tyrosinase gene and the recessive white mutation in chickens. BMC Genomics. 2006; 7: 19. Published online 2006 February 5. doi: 10.1186/1471-2164-7-19.
- Koga A, Wakamatsu Y, Kurosawa J, Hori H.: Oculocutaneous albinism in the i6 mutant of the medaka fish is associated with a deletion in the tyrosinase gene. Pigment Cell Res. 1999 Aug;12(4):252-8. PMID: 10454293
- Cook D, Brooks S, Bellone R, Bailey E.: Missense Mutation in Exon 2 of SLC36A1 Responsible for Champagne Dilution in Horses. PLoS Genet. 2008 Sep 19;4(9):e1000195. PMID 18802473
Verhaltensforschung
- Vitus B. Dröscher: Weiße Löwen müssen sterben. Spielregeln der Macht im Tierreich. Rasch und Röhring, Hamburg 1989, S. 212-244 (Mobbing: „Tötet den Außenseiter!“)
- Irenäus Eibl-Eibesfeldt: Die Biologie des menschlichen Verhaltens. Piper, München und Zürich 1986, S. 409-417 (Mobbing: „Bewahrung der Gruppenidentät“)
Einzelnachweise
- ↑ Friedrich Kluge, Elmar Seebold: Etymologisches Wörterbuch der deutschen Sprache. 2002 Walter de Gruyter, ISBN 3110174731
- ↑ Grønskov K, Ek J, Brondum-Nielsen K: Oculocutaneous albinism. Orphanet J Rare Dis. 2007 Nov 2;2:43. PMID 17980020
- ↑ http://dasmagazin.ch/index.php/die-gejagten/ Das Magazin: Die Gejagten
- ↑ IRIN News: Senegal: Albinos face perilous social rejection
- ↑ BBC News: Albinos hit by Zimbabwe's race divide
- ↑ Akihiko Koga, Hidehito Inagaki, Yoshitaka Bessho and Hiroshi Hori: Insertion of a novel transposable element in the tyrosinase gene is responsible for an albino mutation in the medaka fish, Oryzias latipes. Molecular and General Genetics MGG Volume 249, Number 4 / July, 1995 S.400-405 DOI 10.1007/BF00287101 ISSN 0026-8925 (Print) 1432-1874 (Online)
- ↑ Koga A, Wakamatsu Y, Kurosawa J, Hori H: Oculocutaneous albinism in the i6 mutant of the medaka fish is associated with a deletion in the tyrosinase gene. Pigment Cell Res. 1999 Aug;12(4):252-8. PMID 10454293
- ↑ Koga A, Hori H.: Albinism due to transposable element insertion in fish. Pigment Cell Res. 1997 Dec;10(6):377-81. Review. PMID 8552044
- ↑ Iida A, Inagaki H, Suzuki M, Wakamatsu Y, Hori H, Koga A: The tyrosinase gene of the i(b) albino mutant of the medaka fish carries a transposable element insertion in the promoter region. Pigment Cell Res. 2004 Apr;17(2):158-64.Click here to read PMID 15016305
- ↑ Protas ME, Hersey C, Kochanek D, Zhou Y, Wilkens H, Jeffery WR, Zon LI, Borowsky R, Tabin CJ: Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nat Genet. 2006 Jan;38(1):107-11. Epub 2005 Dec 11. PMID 16341223
- ↑ Kobayashi T, Hearing VJ: Direct interaction of tyrosinase with Tyrp1 to form heterodimeric complexes in vivo. In: J. Cell. Sci.. 120, Nr. Pt 24, December 2007, S. 4261–8. doi:10.1242/jcs.017913. PMID 18042623
- ↑ Adam Amsterdam, Shawn Burgess, Gregory Golling, Wenbiao Chen, Zhaoxia Sun, Karen Townsend, Sarah Farrington, Maryann Haldi und Nancy Hopkins: A large-scale insertional mutagenesis screen in zebrafish. Genes & Development 1999. 13: 2713-2724
- ↑ Meredith Protas, Melissa Conrad, Joshua B. Gross, Clifford Tabin und Richard Borowsky: Regressive Evolution in the Mexican Cave Tetra, Astyanax mexicanus. Curr Biol. 2007 March 6; 17(5): 452–454. DOI 10.1016/j.cub.20
- ↑ R. Borowsky, and H. Wilkens: Mapping a Cave Fish Genome: Polygenic Systems and Regressive Evolution. J Hered. 2002 Jan-Feb;93(1):19-21.Click here to read. PMID 12011170
Weblinks
- NOAH Albinismus Selbsthilfegruppe e.V.
- Die Seite rund um die medizinischen Aspekte des Albinismus
- Weltspiegel-Beitrag: Mörderjagd auf Albinos WDR, Sonntag, 14. September 2008

Bitte beachte den Hinweis zu Gesundheitsthemen!














Wikimedia Foundation.