- Pleomorphes Adenom
-
Klassifikation nach ICD-10 D11.0 Pleomorphes Adenom der Glandula parotis ICD-10 online (WHO-Version 2011) Das pleomorphe Adenom ist ein gutartiger Speicheldrüsentumor, der sich meist im Bereich der Ohrspeicheldrüse, seltener in anderen Speicheldrüsen manifestiert. Mit einem Anteil von 85 % ist es die häufigste benigne Neoplasie der Speicheldrüsen und macht etwa die Hälfte aller Speicheldrüsentumoren insgesamt aus.[1][2]
Pleomorphe Adenome wachsen langsam und bleiben lange asymptomatisch. Therapie der Wahl ist die chirurgische Entfernung des Tumors, mit der sich eine definitive Heilung erzielen lässt. Verbleiben allerdings Tumorreste im Körper, so können hartnäckige Rezidive die Folge sein. Darüber hinaus kann es zur Entstehung eines malignen Tumors innerhalb eines pleomorphen Adenoms kommen, wobei das Risiko zu Anfang gering ist und erst mit der Zeit wächst.[3]
Inhaltsverzeichnis
Epidemiologie
Der Altersgipfel liegt in der 4.–6. Lebensdekade, das mittlere Manifestationsalter liegt bei 43 Jahren. Frauen sind etwas häufiger betroffen als Männer (3:2). Weiße erkranken häufiger als Angehörige anderer Rassen.[1]
Ätiologie
Die der Tumorentstehung zugrunde liegenden Ursachen sind unbekannt. Anders als beim Warthin-Tumor, dem zweithäufigsten Tumor der Ohrspeicheldrüse, besteht offenbar keine Assoziation zum Zigarettenrauchen.[4]
Molekulargenetisch bedeutsam für die Entstehung pleomorpher Adenome ist eine Translokation und Überexpression der für Transkriptionsfaktoren kodierenden Gene PLAG (Chromosom 8q) und HMGA2 (Chromosom 12q).[3]
Pathologie
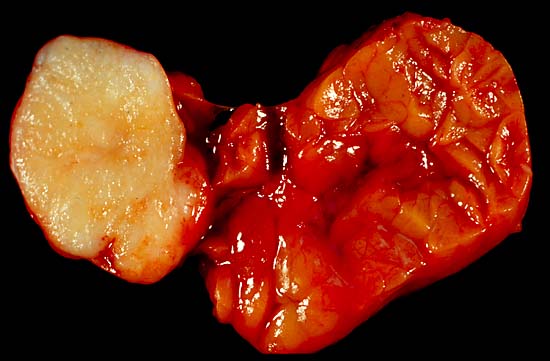
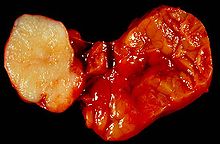 Pleomorphes Adenom der Glandula submandibularis.
Pleomorphes Adenom der Glandula submandibularis.
Hauptlokalisation des pleomorphen Adenoms ist die Ohrspeicheldrüse (85 %), gefolgt von den kleinen Mundspeicheldrüsen (10 %) und der Unterkieferspeicheldrüse (5 %).[5] Seltenere Manifestationsorte sind die Unterzungenspeicheldrüse, Tränendrüse[6], das Tracheobronchialsystem[7], die Nasenhöhle[8], Haut, Brustdrüse[9] oder das Weichgewebe.[3]
Innerhalb der Ohrspeicheldrüse betrifft der Tumor meist den oberflächlichen (75 %), seltener den tiefen (25 %) Drüsenlappen.[10]
Makroskopisch imponiert der Tumor als wenige Millimeter bis mehrere Zentimeter große, gut umschriebene, gummiartig-elastische oder fleischige, grau-weiße Gewebsmasse mit mukoider oder glänzender Schnittfläche. Rezidivtumoren sind häufig mehrknotig aufgebaut.
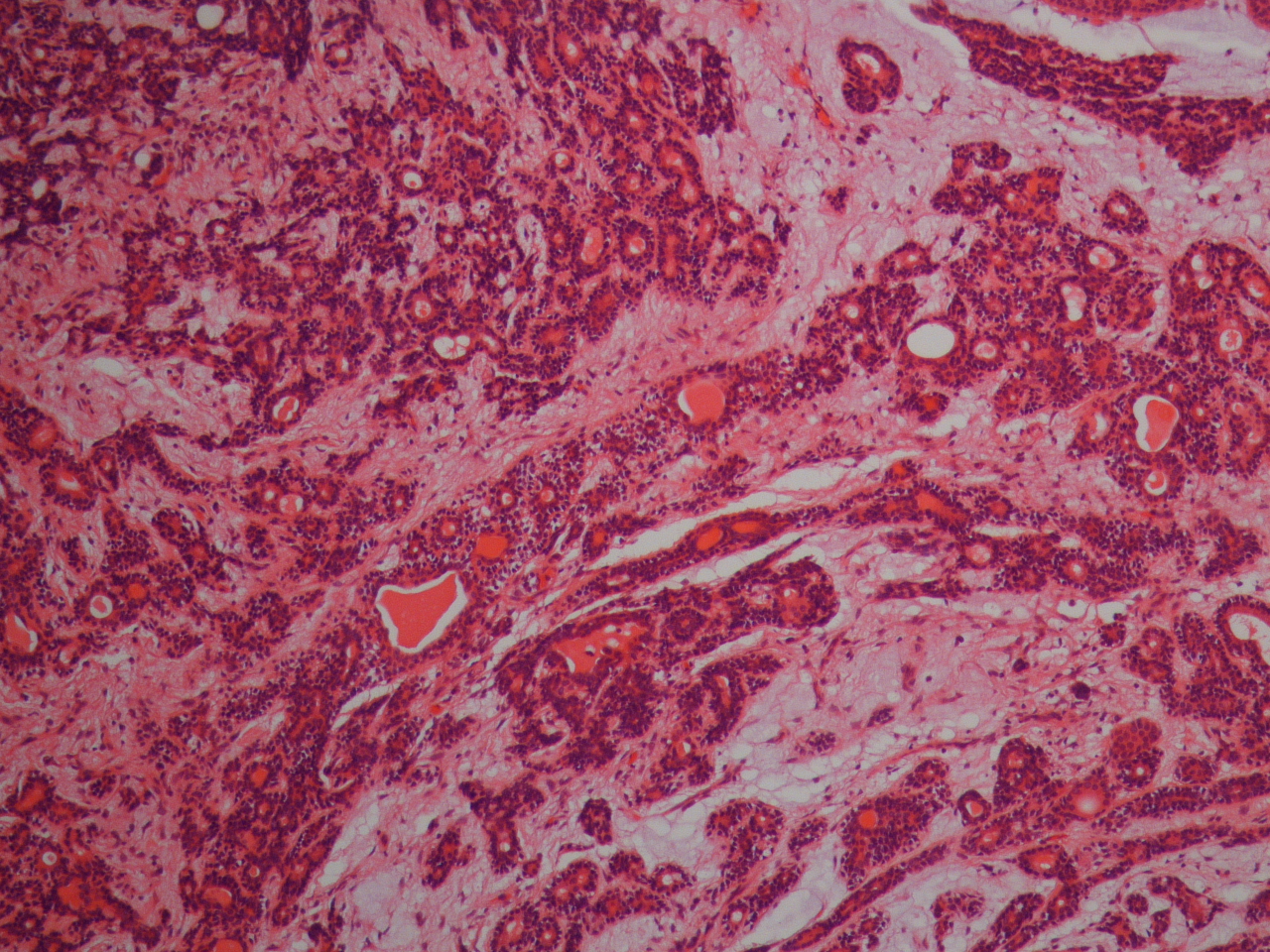
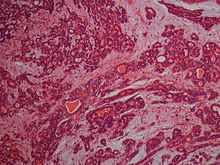 Histologie eines pleomorphen Adenoms der Ohrspeicheldrüse.
Histologie eines pleomorphen Adenoms der Ohrspeicheldrüse.Histologisch zeigt sich ein häufig durch eine dünne bindegewebige Kapsel begrenzter Tumor mit vielgestaltiger Histoarchitektur, der sich aus epithelialen und modifizierten myoepithelialen Zellen zusammensetzt. Diese bilden strangförmige, gangartige und solide Verbände oder sind bienenschwarmartig innerhalb eines wechselnd umfangreichen mukoiden, myxoiden oder chondroiden Stromas verteilt.[2]
Immunhistochemie
Die epitheliale Komponente des Tumors zeigt eine Positivität für Carcinoembryonales Antigen (CEA), das epitheliale Membranantigen (EMA), c-kit und in wechselndem Ausmaß auch für S-100. Verlässliche Marker der myoepithelialen Komponente sind insbesondere p63 und Calponin; daneben besteht eine Positivität für Zytokeratine, S-100 oder GFAP.
Hilfreich bei der differentialdiagnostischen Abgrenzung vom adenoid-zystischen Karzinom ist die Expression der Marker Ki-67 und p53. Während diese beim pleomorphen Adenom im Mittel von 1,6 beziehungsweise 1,2 Prozent der Tumorzellen exprimiert werden, sind im Falle des adenoid-zystischen Karzinoms 20–55 bzw. 4–24 Prozent der Tumorzellen positiv. Darüber hinaus zeigen adenoid-zystische Karzinome eine wesentlich stärkere Expression des Markers bcl-2.[3]
Diagnose
Pleomorphe Adenome werden üblicherweise als langsam wachsende, feste Resistenz klinisch auffällig. Dabei sind die Tumoren im Übrigen weitgehend symptomlos und werden nicht selten zufällig im Rahmen von Routineuntersuchungen entdeckt. Häufig bestehen die Läsionen bereits seit einem Jahr oder länger, bevor der Patient den Arzt aufsucht.
Die Ausdehnung des Befundes kann mittels bildgebender Verfahren (Magnetresonanztomographie, Computertomographie oder Sonographie) ermittelt werden. Eine definitive Diagnose wird vom Pathologen durch histologische Untersuchung des Operationspräparates oder manchmal auch bereits anhand von bei einer präoperativ durchgeführten Feinnadelpunktion gewonnenem Zellmaterial gestellt.[1]
Differentialdiagnose
Klinische Differentialdiagnosen sind der Warthin-Tumor und weitere Speicheldrüsentumoren, eine Sarkoidose, ein Lymphom oder eine Lymphadenopathie.[1]
Im Rahmen der histologischen Diagnostik auszuschließende Tumoren sind insbesondere monomorphe Adenome (Basalzelladenom, Myoepitheliom), das adenoid-zystische Karzinom, das polymorphe niedriggradige Adenokarzinom, das mukoepidermoide Karzinom sowie verschiedene mesenchymale Neoplasien.[3]
Therapie
Therapie der Wahl ist die vollständige chirurgische Entfernung des Befundes. Im Falle eines Befalls der Ohrspeicheldrüse führt man meist eine Teilentfernung der Drüse, seltener lediglich eine Ausschälung des Tumorgewebes (Enukleation) durch, um den Gesichtsnerv (N.facialis, N.VII) möglichst zu schonen.[1]
Prognose
Bei üblichem chirurgischen Vorgehen werden Rezidivraten von 1–5 Prozent beschrieben. Erfolgt lediglich eine Ausschälung (Enukleation) des Befundes, so ist ein Wiederauftreten des Tumors in bis zu 50 Prozent der Fälle zu beobachten.[1] Stromareiche Tumoren neigen offenbar verstärkt zu Rezidiven, mutmaßlich, weil es unter der Operation eher zu einer Verschleppung des oft weichen Stromas kommt. Hingegen sind zellreiche Tumoren möglicherweise mit einem erhöhten Entartungsrisiko behaftet. Das Risiko der Entstehung eines malignen Tumors in einem pleomorphen Adenom (Karzinom ex pleomorphes Adenom) wird verschiedenen Studien zufolge mit 2–23 Prozent, im Mittel mit 6 Prozent angegeben, wobei die Entartungsfrequenz umso höher ist, je länger der Befund besteht.
Auch eine Metastasierung des eigentlich benignen Tumors wird beobachtet. Da diese häufig erst nach langjährigen Verläufen mit multiplen Operationen beobachtet werden und die Metastasen häufig wie auch der Primärtumor keine Malignitätskriterien erkennen lassen, geht man davon aus, dass die Verschleppung von Tumorgewebe unter der Operation, etwa über eröffnete Gefäße, eine Rolle spielen könnte. Tumorabsiedlungen finden sich in absteigender Häufigkeit in Knochen (50 Prozent), Lunge (30 Prozent) und Lymphknoten (30 Prozent) sowie selten an anderen Lokalisationen wie Kopfhaut, Bauchwand oder Leber.[3]
Einzelnachweise
- ↑ a b c d e f Wagner AL, Haag J. Parotid, Pleomorphic Adenoma (18. April 2007); http://emedicine.medscape.com/article/384327-overview
- ↑ a b Riede U, Werner M, Schäfer H. Allgemeine und spezielle Pathologie, 5. Auflage, Thieme, 2004. ISBN 3-13683305-8
- ↑ a b c d e f Fletcher CDM. Histopathology of Tumors, 3rd edition, Churchill Livingston Elsevier, 2007. ISBN 0-443-07434-8
- ↑ Sadetzki S, Oberman B, Mandelzweig L, Chetrit A, Ben-Tal T, Jarus-Hakak A, Duvdevani S, Cardis E, Wolf M. Smoking and risk of parotid gland tumors: a nationwide case-control study. Cancer. 2008 May 1;112(9):1974-82. PMID 18361448
- ↑ Voz ML, Van de Ven WJ, Kas K. First insights into the molecular basis of pleomorphic adenomas of the salivary glands. Adv Dent Res. 2000 Dec;14:81-3. PMID 11842929
- ↑ Marshall AF, White DR, Shockley WW. Pleomorphic adenoma in the palpebral lobe of the lacrimal gland. Arch Pathol Lab Med. 2004 Dec;128(12):1385-94. PMID 15578883
- ↑ Gaissert HA, Mark EJ. Tracheobronchial gland tumors. Cancer Control. 2006 Oct;13(4):286-94. PMID 17075566
- ↑ Karakus MF, Ozcan KM, Dere H. Endoscopic resection of pleomorphic adenoma of the nasal septum. Tumori. 2007 May-Jun;93(3):300-1. PMID 17679469
- ↑ Sato K, Ueda Y, Shimasaki M, Ozaki M, Nitta N, Chada K, Ishikawa Y, Katsuda S. Pleomorphic adenoma (benign mixed tumor) of the breast: a case report and review of the literature. Pathol Res Pract. 2005;201(4):333-9. PMID 15991841
- ↑ PathConsult: Benign Mixed Tumor (6. Januar 2006), Elsevier; http://www.pathconsultddx.com/pathCon/diagnosis?TXTBOX2=pleo&pii=S1559-8675%2806%2970117-6

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.