- Louis-Bar-Syndrom
-
Klassifikation nach ICD-10 G11.3 Zerebellare Ataxie mit defektem DNA-Reparatursystem ICD-10 online (WHO-Version 2011) 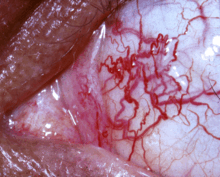 Bindehaut-Teleangiektasien
Bindehaut-Teleangiektasien
Das Louis-Bar-Syndrom (auch Ataxia teleangiectatica (Ataxia teleangiectasia) oder Boder-Sedgwick-Syndrom) ist eine vererbte Systemerkrankung und wird zu den Phakomatosen und Chromosomenbruchsyndromen gezählt. In der neurologischen Systematik wird sie zu den Heredoataxien (vererbte Ataxien) gerechnet. Benannt wurde die Krankheit nach der belgischen Ärztin Denise Louis-Bar.
Inhaltsverzeichnis
Epidemiologie
Der Erbgang ist autosomal-rezessiv und betrifft das sogenannte AT-Gen auf Chromosom 11 Genlocus q23. Das mutierte Gen wird als ATM bezeichnet. 0,5 bis 1 % der Bevölkerung sind gesunde (heterozygote) Träger der Mutation. Etwa eins von 40.000 Neugeborenen ist für das defekte Gen homozygot und wird an der Ataxia teleangiectatica erkranken.
Pathophysiologie
Das ATM-Gen codiert für die Serin-Proteinkinase ATM, die als Sensor von DNA-Schäden durch UV-Strahlung und als Regulator von DNA-Reparaturvorgängen oder programmiertem Zelltod (Apoptose) eine Rolle spielt. Aus der Tatsache, dass viele Zelllinien von der Mutation betroffen sind, erklärt sich die Vielfalt der Symptome des Louis-Bar-Syndroms.
Symptomatik
Die ersten Symptome treten um das zweite bis dritte Lebensjahr auf. Kennzeichnend sind eine zerebelläre Ataxie (u. a. Gang- und Standunsicherheit) mit Kleinhirnatrophie (Substanzschwund) vor allem im Bereich des Vermis, Athetose (dystone Bewegungsstörung), Störungen der Augenbewegungen sowie ein physischer und später auch psychischer Entwicklungsrückstand. Außerdem treten Teleangiektasien (Erweiterungen der kleinen Arterien) vor allem im Gesicht und auf der Bindehaut des Auges auf und die betroffenen Kinder haben durch einen T-Zell-Defekt eine verminderte Immunkompetenz, so dass sie zu Infekten neigen und weitaus häufiger als die Normalbevölkerung Leukämien und Hodgkin-Lymphome entwickeln. Die Krebsrate soll um den Faktor 100 höher liegen als in der Normalpopulation. Erkrankte haben daher auch eine erhöhte Empfindlichkeit gegenüber ionisierender Strahlung und sollten möglichst wenig geröntgt werden. Weitere häufige Symptome sind Hypersalivation (Speichelfluss) und Hypogonadismus (Reifestörung der Keimdrüsen).
Diagnose und Behandlung
Die Diagnose wird oft bereits klinisch gestellt. Bestätigung können eine durch Bildgebung nachgewiesene Kleinhirnatrophie und die verminderten Immunglobulin-Werte sowie die Lymphopenie geben. Die Alpha-1-Fetoprotein-Werte sind oft erhöht.
Eine kausale Behandlung ist derzeit nicht möglich. Es werden vor allem die pulmonalen Infekte mit Antibiotika therapiert. Wichtig ist die konsequente Impfung der Kinder, wobei Lebendimpfstoffe absolut kontraindiziert sind.
Prognose
Die Lebenserwartung ist aufgrund rekurrierender Lungenentzündungen und der erhöhten Krebsrate deutlich reduziert. In der Literatur wird die mediane Lebenserwartung mit ca. 20 Jahren beziffert. Es sind Fälle bekannt, in denen Menschen mit diesem Syndrom das zwanzigste Lebensjahr weit überschritten haben, abhängig von Schwere und Verlauf der Krankheit.
Quellen
- Wallesch (Hrsg.): Neurologie. Diagnostik und Therapie in Klinik und Praxis. Elsevier Urban & Fischer, 2005. ISBN 3-437-23390-4.

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Erbkrankheit
- Krankheitsbild in der Kinderheilkunde
- Krankheitsbild in der Neurologie
Wikimedia Foundation.