- Roberts-Syndrom
-
Klassifikation nach ICD-10 Q73.8 Sonstige Reduktionsdefekte nicht näher bezeichneter Extremität(en) ICD-10 online (WHO-Version 2011) 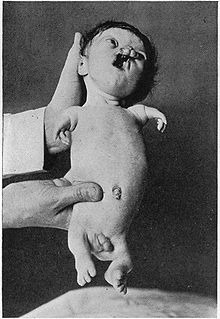 Ein Neugeborenes mit Tetraphokomelie einer Ausprägung des Roberts-Syndroms. Das Kind starb 1919 im Alter von 15 Tagen.
Ein Neugeborenes mit Tetraphokomelie einer Ausprägung des Roberts-Syndroms. Das Kind starb 1919 im Alter von 15 Tagen.
Das Roberts-Syndrom, auch Pseudothalidomid-Syndrom, Appelt-Gerken-Lenz-Syndrom oder Roberts-SC-Phokomelie genannt, ist eine sehr seltene autosomal-rezessiv vererbte schwerwiegende Fehlbildung.
Inhaltsverzeichnis
Klinisches Bild
Die vom Roberts-Syndrom betroffenen Patienten weisen eine Phokomelie (meist eine Tetraphokomelie, das heißt das Fehlen aller vier Gliedmaßen) auf, das Wachstum ist vor- und nachgeburtlich verzögert und durch eine kraniofaziale Fehlbildungen und verschiedene weitere Fehlbildungen gekennzeichnet. Dazu gehören beispielsweise Lippen-Kiefer-Gaumenspalte, Mikrozephalie mit Brachycephalie. Des Weiteren werden in vielen Fällen Missbildungen der Nieren, Herzfehler, Hyperplasie des Penis oder der Klitoris und verschiedene Anomalien der Augen, wie beispielsweise eine Trübung der Hornhaut beobachtet. Überlebende Kinder haben sehr häufig eine geistige Behinderung.[1]
Die Bezeichnung Pseudothalidomid-Syndrom ist auf die bei vielen Contergan-Opfern zu beobachtenden, dem Roberts-Syndrom sehr ähnlichen, Fehlbildungen (insbesondere Tetraphokomelie) zurückzuführen.
Epidemiologie
Das Roberts-Syndrom ist eine sehr seltene Erkrankung. Weltweit wurden bisher etwa 150 Fälle berichtet.
Ätiologie und Genetik
Die Ursache das Roberts-Syndrom sind Mutationen im ESCO2-Gen, das sich beim Menschen auf Chromosom 8 Genlocus p21.1 befindet. Das Genprodukt von ESCO2, eine N-Acetyltransferase, die beim Menschen aus 601 Aminosäuren besteht,[2] spielt bei der Zellteilung in der S-Phase bei der Verdopplung der Chromatiden eine wichtige Rolle.[3] Die Trägerfrequenz der Mutation auf dem ESCO2-Gen ist unbekannt. Das ESCO2-Gen besteht aus elf Exons mit 30,3 kb.
Behandlung und Prognose
Die meisten Kinder mit Roberts-Syndrom sind Totgeburten oder sterben in einer sehr frühen Phase nach der Geburt. Es sind jedoch auch Fälle bekannt, in denen sich betroffene Kinder geistig normal entwickelten. Die Behandlung der Patienten ist individuell sehr unterschiedlich und zielt im Wesentlichen auf korrigierende Maßnahmen aus den verschiedensten medizinischen Bereichen ab, um die Lebensqualität zu verbessern. Zu den Maßnahmen gehören unter anderem chirurgische Eingriffe kosmetischer und rekonstruktiver Art, beispielsweise bei einer Lippen-Kiefer-Gaumenspalte und frühzeitige handchirurgische Korrekturen, um die Entwicklung des Greifens zu fördern. Herzfehler und Nierenfunktionsstörungen werden symptomatisch behandelt.
Erstbeschreibung
Namensgeber des Roberts-Syndroms ist der US-amerikanische Chirurg John Bingham Roberts (1852–1924),[4] der dieses Syndrom erstmals 1919 wissenschaftlich beschrieb.[5] 1966 beschrieben die deutschen Genetiker Hans Appelt, Hartmut Gerken und Widukind Lenz einen Fall des inzwischen weitgehend in Vergessenheit geratenen Syndroms,[6] weshalb man auch den synonymen Begriff Appelt-Gerken-Lenz-Syndrom für das Roberts-Syndrom verwendet.
Einzelnachweise
- ↑ D. A. Musfeld, E. M. Bühler, S. Heinzl: Roberts-SC-Phokomelie- oder Pseudothalidomid-Syndrom. In: Gynäkologisch-geburtshilfliche Rundschau Band 41, Nummer 1, 2001, S. 3–7, doi:10.1159/000049454
- ↑ UniProt: Q56NI9 (ESCO2 HUMAN). Abgerufen am 12. Januar 2011
- ↑ Roberts-Syndrom bei Online Mendelian Inheritance in Man
- ↑ J. L. Stone: John Bingham Roberts and the first American monograph on human brain surgery. In: Neurosurgery Band 49, Nummer 4, Oktober 2001, S. 974–984, ISSN 0148-396X. PMID 11564261.
- ↑ J. B. Roberts: A child with double cleft of lip and palate, protrusion of the intermaxillary portion of the upper jaw and imperfect development of the bones of the four extremities. In: Ann Surg. Band 70, 1919, S. 252–254.
- ↑ H. Appelt, H. Gerken und W. Lenz: Tetraphokomelie mit Lippen-Kiefer-Gaumenspalte und Clitorishypertrophie - Ein Syndrom. In: Paediatr Paedol. Band 2, 1966, S. 119.
Weiterführende Literatur
- Review-Artikel
- M. Mordillo, H. Vega, E. W. Jabs: Roberts Syndrome. In: R. A. Pagon u.a. (Herausgeber): GeneReviews. University of Washington, Seattle, 1993-2006, PMID 20301332
- S. A. Temtamy, S. Ismail, N. A. Helmy: Roberts syndrome: study of 4 new Rgyptian cases with comparison of clinical and cytogenetic findings. In: Genetic counseling (Geneva, Switzerland) Band 17, Nummer 1, 2006, S. 1–13, ISSN 1015-8146. PMID 16719272. (Review).
- M. Urban, C. Opitz u.a.: Bilaterally cleft lip, limb defects, and haematological manifestations: Roberts syndrome versus TAR syndrome. In: American journal of medical genetics Band 79, Nummer 3, September 1998, S. 155–160, ISSN 0148-7299. PMID 9788553. (Review).
- A. K. Sinha, R. S. Verma, V. J. Mani: Clinical heterogeneity of skeletal dysplasia in Roberts syndrome: a review. In: Human heredity Band 44, Nummer 3, 1994 May-Jun, S. 121–126, ISSN 0001-5652. PMID 8039795. (Review).
- D. J. Van Den Berg, U. Francke: Roberts syndrome: a review of 100 cases and a new rating system for severity. In: American journal of medical genetics Band 47, Nummer 7, November 1993, S. 1104–1123, ISSN 0148-7299. doi:10.1002/ajmg.1320470735. PMID 8291532. (Review).
- P. Iannetti, C. E. Schwartz u.a.: Norman-Roberts syndrome: clinical and molecular studies. In: American journal of medical genetics Band 47, Nummer 1, August 1993, S. 95–99, ISSN 0148-7299. doi:10.1002/ajmg.1320470120. PMID 8368261. (Review).
- Originalforschung
- A. Al Kaissi, R. Csepan u.a.: Femoral-tibial-synostosis in a child with Roberts syndrome (Pseudothalidomide): a case report. In: Cases journal Band 1, Nummer 1, 2008, S. 109, ISSN 1757-1626. doi:10.1186/1757-1626-1-109. PMID 18710560. PMC PMC2542345. (Open Access)
- J. Khandrani u. a.: Anesthetic care of Roberts syndrome: A Case report. In: The Internet Journal of Anesthesiology ISSN 1092-406X Band 24, Nummer 2, 2010
Weblinks
- Roberts syndrome (englisch)
- Roberts' pseudothalidomide syndrome Who named it? (englisch)
- John Bingham Roberts Who named it? (englisch)
Kategorien:- Fehlbildung
- Erbkrankheit
- Krankheitsbild in der Kinderheilkunde
Wikimedia Foundation.