- Paroxysmale nokturne Hämoglobinurie
-
Bei der paroxysmalen nächtlichen Hämoglobinurie (PNH) handelt es sich um eine seltene erworbene Erkrankung blutbildender Stammzellen. Ursache ist eine Genmutation, welche die Funktion gewisser Proteine auf den Blutzellen stört. Kardinalsymptome der Krankheit sind eine Hämolyse, eine verstärkte Blutgerinnungsneigung und Blutarmut und gegebenenfalls Verminderung der weißen Blutkörperchen. Charakteristisch sind hämolyische Anfälle (Paroxysmen) mit einer erhöhten Nachweisbarkeit des Hämoglobins im Morgenurin.
Synonym wird auch von einer Marchiafava-Micheli-Anämie oder besser einem Marchiafava-Micheli-Syndrom gesprochen, da alle Linien der Blutzellen betroffen sein können.
Erstmalig beschrieben wurde die Erkrankung 1882 von Struebing [1].
Inhaltsverzeichnis
Pathogenese und Epidemiologie
Der PNH liegt eine somatische Genmutation zu Grunde. Dabei wird durch diese Mutation in blutbildenden Stammzellen, während des Lebens eines Menschen, das PIG-A-Gen in seiner Funktion eingeschränkt oder vollends behindert. Dieses Gen kodiert für das Enzym N-Acetylglukosaminyltransferase, welches für die Bildung sogenannter GPI-Anker vonnöten ist. Diese GPI-Anker verankern Proteine in der Zellmembran von Zellen. Fehlt diese Verankerung, oder sind die Anker defekt gebildet, kommt es zum Funktionsverlust der mit ihnen verbundenen Proteine auf den reifen, von der mutierten Stammzelle abstammenden Blutzelle. Dadurch bedingt können die Komplement-Regulationsproteine CD55 u. CD59 nicht an die roten Blutkörperchen andocken, und es kommt zur intravasalen Hämolyse. Da die Mutation nicht an den Keimzellen auftritt ist sie nicht vererbbar. Eine Häufung unter Verwandten oder nach Geschlecht wurde bisher nicht beobachtet. Die Rate der Neuerkrankungen wird zwischen 1 : 100.000 und 1 : 500.000 pro Jahr beziffert. Gehäuft bricht die Krankheit im Alter von 25 bis 45 Jahren aus.[2]
Symptome
Klinisch im Vordergrund steht bei der PNH die Hämolyse. Es sind zwei GPI-verankerte Oberflächenproteine auf Erythrozyten bekannt, die komplementhemmend wirken (CD 55, CD 59) und deren Funktion auf dem mutierten Zellklon gestört sind. Dadurch greift das im Blut vorhandene Komplementsystem die betreffenden roten Blutkörperchen an und zerstört sie. Der Anteil der im Blut befindlichen mutierten Zellen schwankt dabei im Verlauf der Erkrankung. So kommt es zu anfallsartigen hämolytischen Krisen, wenn viele defekte Zellen gebildet wurden. Begleitet wird die Erkrankung durch eine Neigung zur Bildung von Thrombosen. Die Blutgerinnsel im Rahmen der PNH treten vorwiegend in Venen der Bauchorgane, des Gehirns oder der Körperperipherie auf. Rund die Hälfte der Erkrankten entwickeln eine Thrombose, die in vielen Fällen tödlich verläuft.
Begleitet werden die hämolytischen Anfälle oft durch Bauchkrämpfe, Kopfschmerzen, Rückenschmerzen und Potenzstörungen. Die Schmerzzustände werden auf kleinste Thromben im Zuge de erhöhten Blutgerinnungsneigung zurückgeführt. Als mögliche Erklärung für die Krämpfe und Potenzbeeinträchtigung gilt die Bindung von NO an das aus zerstörten Erythrozyten freigewordene Hämoglobin. NO entspannt die glatte Muskulatur und seine Bindung führt im Umkehrschluss zu erhöhter Spannung der glatten Muskulatur in Hohlorganen und Gefäßen. Das besonders im Morgenurin besonders stark ausgeschiedene Hämoglobin hat der Krankheit jedenfalls ihren Namen gegeben. Es ist neben der aus der Hämolyse resultierenden Anämie meist erstes klinisch fassbares Zeichen der Krankheit.
Diese Blutarmut kann sich auf eine Linie der Blutzellen (z. B. nur rote Blutkörperchen), oder auf mehrere beziehen. Bei besonders schwerem Bild sind alle Zellreihen betroffen (Panzytopenie). Sind Immunzellen des Blutes von der Mutation betroffen, so kommt es auch zu einer Schwächung des Immunsystems. Des Weiteren ist die PNH oft mit dem Bild der Aplastische Anämie oder anderen Knochenmarkserkrankungen verknüpft.[3]Diagnose
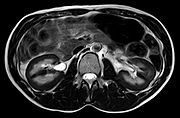 MRT: Signalarme Darstellung der Nieren durch Eisenablagerungen bei Marchiafava-Syndrom.
MRT: Signalarme Darstellung der Nieren durch Eisenablagerungen bei Marchiafava-Syndrom.Die Ausprägung der Symptome sind von Patient zu Patient sehr unterschiedlich. In manchen Fällen liegen auch keine Symptome vor, man spricht hierbei von einer subklinischen PNH. Klassischerweise findet im Blut von Patienten mit PNH eine Anämie, ggf. kombiniert mit einer Thrombozytopenie und Neutropenie. Als laborchemische Zeichen der Hämolyse findet man eine Erhöhung der Lactatdehydrogenase und des indirekten Bilirubins sowie eine Erniedrigung des Haptoglobins. Der Coombs-Test ist bei der PNH in der Regel negativ. Methode der Wahl zur Diagnosesicherung ist die Durchflusszytometrie. Mit dieser Methode wird das Fehlen der GPI-Anker-abhängigen Proteine nachgewiesen. Der Nachweis zweier fehlender Proteine auf zwei verschiedenen Zellreihen des Blutes gilt als beweisend für eine PNH. Eine neue Methode zur Diagnostik bietet Aerolysin. Es handelt sich hierbei um ein Toxin des Bakteriums Aeromonas hydrophilius, das unmittelbar an GPI-Anker bindet. Es besteht die Möglichkeit mit einem Fluoreszenzfarbstoff-kombiniertem Aerolysin das Fehlen der GPI-Anker nachzuweisen. Des Weiteren steht auch die Möglichkeit durch molekularbiologische Verfahren (z.B. PCR) direkt die Genmutation in betreffenden Zellen nachzuweisen. Aufgrund des technischen Aufwands werden aber in der Regel einfachere Methoden bevorzugt.[4]
Prognose
Die durchschnittliche Überlebenszeit der Patienten nach Diagnosestellung beträgt zwischen zehn und fünfzehn Jahren. In rund 15% der Fälle wurde eine Spontanheilung beobachtet. Ein Übergang in eine Aplastische Anämie (auch umgekehrt) oder eine Akute myeloische Leukämie ist allerdings möglich. [5]
Therapie
Bei den meisten Patienten steht eine Therapie der Symptome im Vordergrund. Die Blutarmut kann in der Regel durch Transfusionen von Erythrozytenkonzentraten ausgeglichen werden. Falls durch die Hämolyse ein Eisenmangel entsteht sollte Eisen in Tablettenform verabreicht werden. Des Weiteren ist die Gabe von Folsäure empfohlen, da die Neubildung zerstörter Blutzellen zu einem erhöhten Bedarf von Folsäure führt. Da Infekte als ein Faktor angesehen werden, der hämolytische Krisen fördert, sollten bei bakteriellen Infektionen frühzeitig Antibiotika gegeben werden. Außerdem ist für eine ausreichende Gabe von Schmerzmitteln bei bestehenden Schmerzzuständen zu sorgen. Die Gabe von Kortikosteroiden zur Unterdrückung der Komplementreaktion gegen Blutzellen ist umstritten, soll aber in einigen Fällen als Kurztherapie während hämolytischen Anfällen dem Patienten Erleichterung verschafft haben. Nach einer aufgetretenen Thrombose, oder einem hohen Anteil von mutierten Blutzellen ist eine dauerhafte Gerinnungshemmung durch Cumarine angezeigt. Für eine kurzzeitige Gerinnungsunterdrückung können Heparine verwandt werden.
Einzige Heilungsmöglichkeit besteht in einer Knochenmarkstransplantation. Da die Überlebenszeit nach diesem Eingriff aber oft unter der normalen Prognose der Krankheit liegt und auch Spontanheilungen auftreten ist dies nur in besonders schweren Fällen eine Möglichkeit.
Ein gentechnisch hergestellter Antikörper gegen den Komplementfaktor C5 wurde in klinischen Studien getestet. Durch diesen Wirkstoff namens Eculizumab soll die Hämolyse unter Kontrolle gehalten werden können.[6]. Eculizumab wurde 2007 in den USA und in der Europäischen Union als Orphan-Arzneimittel unter dem Markennamen Soliris® zugelassen. Durch Eculizumab kann bei der Hälfte der Patienten eine Transfusionsfreiheit erzielt werden. Langzeitergebnisse fehlen aber bisher, sodass Patienten ausschließlich an speziellen Zentren am besten im Rahmen von Studien behandelt werden sollten.Quellen
- ↑ http://www.krebsinfo.de/ki/empfehlung/leu/802_10_PNH.pdf MANUAL Leukämien, myelodysplastische und myeloproliferative Syndrome
- ↑ Alexander Röth, Ulrich Dürsen : Paroxysmale nächtliche Hämoglobinurie , Deutsches Ärzteblatt, Jahrgang 104, Heft 4 vom 26. Januar 2007 S. 193
- ↑ André Tichelli, Richard Herrmann in Heiner Greten (Hrsg.) Innere Medizin, 12. Auflage, Stuttgart, 2005 S. 892 ; Alexander Röth, Ulrich Dürsen : Paroxysmale nächtliche Hämoglobinurie , Deutsches Ärzteblatt, Jahrgang 104, Heft 4 vom 26. Januar 2007 S. 193f
- ↑ Alexander Röth, Ulrich Dürsen : Paroxysmale nächtliche Hämoglobinurie , Deutsches Ärzteblatt, Jahrgang 104, Heft 4 vom 26. Januar 2007 S. 194f
- ↑ André Tichelli, Richard Herrmann in Heiner Greten (Hrsg.) Innere Medizin, 12. Auflage, Stuttgart, 2005 S. 892 ; Alexander Röth, Ulrich Dürsen : Paroxysmale nächtliche Hämoglobinurie , Deutsches Ärzteblatt, Jahrgang 104, Heft 4 vom 26. Januar 2007 S. 195
- ↑ Alexander Röth, Ulrich Dürsen : Paroxysmale nächtliche Hämoglobinurie , Deutsches Ärzteblatt, Jahrgang 104, Heft 4 vom 26. Januar 2007 S. 195-197
Weblinks
- Selbsthilfegruppe Aplastische Anämie - PNH
- Aplastic Anemia & MDS International Foundation

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.