- Phylogenetischer Baum
-
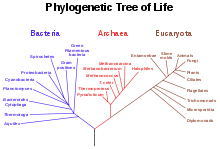 Phylogenetischer Baum basierend auf rRNA Genen
Phylogenetischer Baum basierend auf rRNA Genen
Ein phylogenetischer Baum ist ein Baum, der die evolutionären Beziehungen zwischen verschiedenen Arten oder anderen Einheiten, von denen man vermutet, dass sie einen gemeinsamen Vorfahren besitzen, darstellt. Damit ist ein phylogenetischer Baum eine Form des Kladogramms. In einem phylogenetischen Baum repräsentiert jeder Knoten mit Vorfahren den nächsten gemeinsamen Verwandten dieser Vorfahren. Die Kantenlänge entspricht meist der geschätzten Zeit, in der sich die Arten separiert haben, oder der Anzahl der Mutationen, die während dieser Entwicklung passierten. Jeder Knoten in einem phylogenetischen Baum wird als taxonomische Einheit bezeichnet, wobei man innere Knoten oft als hypothetische taxonomische Einheiten bezeichnet, wenn die entsprechenden Arten oder Einheiten nicht beobachtet werden können.
Inhaltsverzeichnis
Datenquellen und Interpretation
Phylogenetische Bäume werden heute meist anhand von sequenzierten Genen der untersuchten Spezies aufgebaut. Dabei berechnet man ein Sequenzalignment des gleichen Gens (oder evtl. der gleichen Gene) dieser Arten, und verwendet die im Alignment erscheinenden Ähnlichkeiten und Unterschiede, um den Baum aufzubauen. Arten, deren Sequenzen ähnlich sind, liegen im Baum dann wahrscheinlich näher beieinander als solche mit stark unterschiedlichen Sequenzen. Da die Komplexität der Berechnung solcher Bäume jedoch mit der Anzahl der Sequenzen exponentiell ansteigt, verwendet man Heuristiken, um die Bäume zu generieren. Zu den Standardmethoden der molekular-phylogenetischen Baumkonstruktionsmethoden gehören Maximum-Likelihood-, Neighbor-Joining-, Maximum Parsimony- und Bayesische Analyse-Methoden.
Ziel der Erstellung phylogenetischer Bäume ist es, die Evolution möglichst detailliert zu erklären. Allerdings weiß man heute, dass die Gene sich nicht gleichmäßig entwickelt haben. Einige Gene, die heute im Menschen vorkommen, haben beispielsweise nur gemeinsame Vorfahren mit dem Schimpansen, andere kommen in allen Säugetieren vor, etc.
Deshalb können bei der phylogenetischen Analyse verschiedener Gene der gleichen Spezies unterschiedliche phylogenetische Bäume entstehen, die für sich jedoch alle korrekt sind. Um die Entstehungspunkte und Verzweigungen bei der Evolution der einzelnen Arten festzustellen, müssen deshalb verschiedene Genregionen untersucht werden. Weiterhin sollten Ergebnisse aus der klassischen Phylogenie sowie morphologische Merkmale zur Interpretation hinzugezogen werden.
Um diese Probleme in den Griff zu bekommen, werden neuerdings eine große Anzahl von Genen gleichzeitig untersucht; Unregelmäßigkeiten in der Entwicklungsgeschwindigkeit gleichen sich so aus.
Optimal können die Abstammungsverhältnisse erschlossen werden, wenn von allen betrachteten Spezies das gesamte Genom bekannt und seine Gene bestimmt sind. Nach Zuordnung aller zueinander orthologer Gene bleiben diejenigen Gene übrig, in denen sich die Spezies unterscheiden. Phylogenetische Bäume, die auf solchen Betrachtungen der Orthologie beruhen, gelten als die verläßlichsten und stehen für alle sequenzierten Spezies zur Verfügung -- insbesondere die so erstellten Stammbäume der Bakterien und Archäen bieten bereits einen detaillierten Überblick über deren Abstammungsverhältnisse.[1][2]
Wichtige Grundlagenarbeiten zur Konstruktion von phylogenetischen Bäumen wurden Ende der 1960er Jahre von Walter M. Fitch erarbeitet.
Gewurzelte und ungewurzelte Bäume
Ein gewurzelter phylogenetischer Baum ist ein gerichteter Baum mit einem einzelnen Knoten, der dem (meist rechnerisch ermittelten) nächsten gemeinsamen Vorfahren aller Einheiten im Baum entspricht.
Ein ungewurzelter Baum dagegen besitzt keinen ausgezeichneten nächsten gemeinsamen Vorfahren, sondern soll lediglich die Verwandtschaftsnähe oder -ferne der einzelnen Arten darstellen. Näheres zu gewurzelten und ungewurzelten Bäumen ist im Artikel Baum (Graphentheorie) nachzulesen.
Unterschiede zwischen Gen- und Speziesbäumen
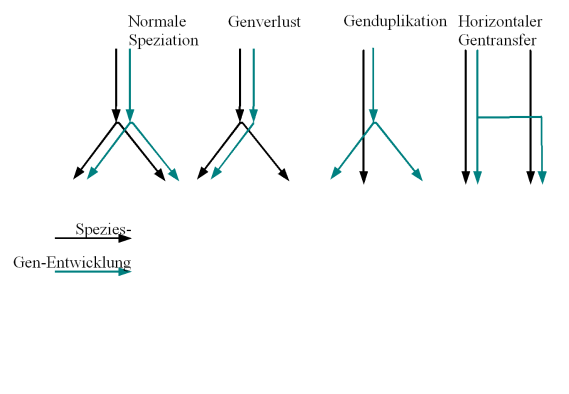
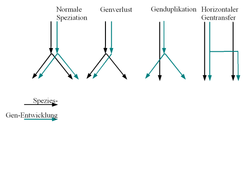 Verschiedene Formen der Gen/Speziesentwicklung
Verschiedene Formen der Gen/SpeziesentwicklungVerschiedene Formen der Gen/Speziesentwicklung: Üblicherweise wird von dem ersten Fall ausgegangen: Speziation geht mit der Aufspaltung der Genentwicklung einher. Eine Rekonstruktion des Artenbaumes wird erschwert durch die drei anderen Fälle:
- das Gen wird nur von einer der neuen Spezies übernommen (Genverlust)
- das Gen wird dupliziert, was bei einer anschließenden Speziation (nicht gezeigt) zu zweideutigen Vergleichsmöglichkeiten führt
- es findet horizontaler Gentransfer zwischen zwei unterschiedlichen Arten statt, die dadurch bei der Baumrekonstruktion fälschlicherweise zusammengerückt werden
→ Siehe auch: Homologie (Biologie), Orthologie, Paralogie.
Methoden
Sequenzanalysen
Übliche Methoden der Bioinformatik zur phylogenetischen Sequenzanalyse sind Parsimony, bei der die geringste Anzahl von "Erklärungen", in diesem Fall Sequenzübereinstimmungen, die Abstammungsverhältnisse klären soll. Beim Neighbour-joining werden alle Sequenzen mit allen in einem Alignment verglichen, die jeweils ähnlichsten zueinander werden als verwandt aufgefasst und in der nächsten Runde des Joinings als eine gemeinsame Art behandelt, bis ein vollständiger Baum entsteht. Gegenwärtig am häufigsten verwendet wird das Maximum Likelihood-Modell, welches auf statistischen Annahmen über die Evolution von Sequenzen beruht.
Orthologieuntersuchungen
Sind viele Genome bekannt und sind die einzelnen Gene darin charakterisiert, wie dies bereits insbesondere bei den Bakterien der Fall ist, können orthologe Gene zwischen Individuen markiert werden. Alles, was nicht ortholog ist, entstammt einer Insertion oder Deletion eines Gens, je nachdem, welche zeitliche Reihenfolge angenommen wird. Damit müssen nur noch solche Ereignisse analysiert werden, um die Abstammungsverhältnisse festzustellen.[3]
Kritik
- Per Definition können phylogenetische Bäume Hybridbildung und lateralen Gentransfer, die ebenfalls wichtige Methoden der Genübertragung sind, nicht darstellen. Deshalb vertreten einige Forscher die Ansicht, dass man nicht einen Baum sondern vielmehr ein phylogenetisches Netz aufbauen sollte (das sich im Sinne der Graphentheorie von einem Baum dadurch unterscheidet, dass es "Querverbindungen" zwischen sonst nicht direkt verwandten Arten zulässt).
- Bäume, die ausgestorbene Arten nicht enthalten, müssen mit Vorsicht interpretiert werden (siehe auch obige Bemerkung zur Interpretation).
Historie der Baum-Metapher
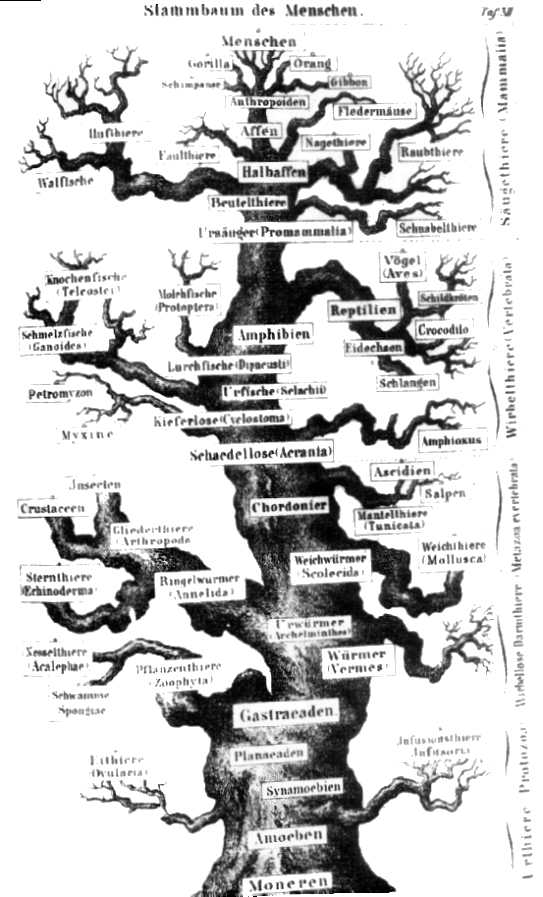
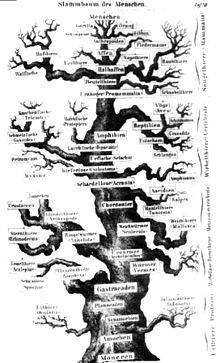 Stammbaum nach Ernst Haeckel (1874)
Stammbaum nach Ernst Haeckel (1874)Die Bezeichnung Baum ist abgeleitet von früheren Vorstellungen des Lebens als Fortschritt von „niedrigeren“ zu „höheren“, komplexeren Formen, wobei die jeweils zugeschriebene Entwicklungshöhe durch die Höhe der Platzierung im „evolutionären Stammbaum“ angedeutet wurde. Als Vorlage für die grafische Darstellung eines solchen Stammbaums diente häufig ein realer Baum.
Eine Veränderung der Stammbaum-Metapher in die eines „Stammflusses“ wurde von Richard Dawkins vorgeschlagen: Man könne das Kladogramm als sich verzweigendes Fluss-System deuten, was u. a. den Vorteil habe, dass diese Metapher keine Höherentwicklung suggeriere.
Literatur
- Walter M. Fitch, Emanuel Margoliash: Construction of phylogenetic trees. In: Science, Band 155, Nr. 3760, 1967, S. 279–284, doi:10.1126/science.155.3760.279, Volltext (PDF)
- Knoop, Volker; Müller, Kai: Gene und Stammbäume - Ein Handbuch zur molekularen Phylogenetik. Spektrum Akademischer Verlag/Heidelberg 2006, ISBN 3-8274-1642-6
- Arndt von Haeseler, Dorit Liebers: Molekulare Evolution, Fischer 2003, ISBN 3-596-15365-4
- Wiesemüller, B., Rothe, H., Henke, W.: Phylogenetische Systematik. Springer/Berlin 2003, ISBN 978-3-540-43643-0
Siehe auch
Einzelnachweise
- ↑ OMAbrowser: Orthology Prediction - Algorithm
- ↑ Status of the OMA Orthologs Project (Enthält aktuelle Bäume)
- ↑ G Fang, Bhardwaj N, Robilotto R, Gerstein MB: Getting Started in Gene Orthology and Functional Analysis. In: PLoS Comput Biol. 6, Nr. 3, 2010, S. e100073. doi:10.1371/journal.pcbi.1000703.
Weblinks
- Tree of Life Web-Projekt (englisch)
- Online-Version eines Phyletischen Baums, erstellt im Rahmen einer diesem Thema gewidmeten Ausgabe der Zeitschrift Science
- Phyletischer Baum von nahezu allen über 4.500 rezenten Säugetierarten, publiziert in Nature, Band 446, 29. März 2007, PMID 17392779)

Wikimedia Foundation.