- Stille-Kupplung
-
Die Stille-Kupplung ist eine Palladium-katalysierte chemische Kupplung einer Organozinnverbindung (Organostannane) mit einem sp2-hybridisierten organischen Halogenid.[1][2] Die Reaktion ist inzwischen in der organischen Synthesechemie sehr weit verbreitet (Zur Hybdrisierung siehe die Geometrie der Valenzstrukturtheorie.
- R-SnR'3 + R"-X ⇌ R-R" + X-SnR'3
X ist typischerweise ein Halogenid, wie Cl, Br, I oder ein Pseudohalogenid wie das Triflat, CF3SO3-.[3][4]
Die Stille-Kupplung wurde 1977 von John Kenneth Stille entdeckt. Stille-Kupplungen waren 1992 in 50% aller veröffentlichten Synthesen, die Kreuz-Kupplungs-Reaktionen enthielten, verwendet worden. Die Reaktion wird zurzeit vor allem im Hinblick auf die Anwendung in der industriellen Synthese von Pharmazeutika weiterentwickelt. Besonders die hohe Toleranz von funktionellen Gruppen macht die Stille-Kupplung attraktiv für die Synthese komplexer Strukturen.
Da Sauerstoff sowohl die Oxidation des Palladium-Katalysators verursacht, als auch eine Homokupplung der Organozinnverbindungen begünstigt, muss die Reaktion unter Inertgas-Atmosphäre und in absolutiertem Lösungsmittel durchgeführt werden.
Als Organozinn-Verbindung nutzt man in der Regel eine Trimethylstannyl- oder Tributylstannylverbindung. Obwohl die Trimethylstannylverbindung, verglichen mit der Tributylverbindung eine höhere Reaktivität zeigt, verwendet man erstere ungern, da sie eine 1000 mal höhere Toxizität zeigt. Daher verwendet man Trimethylstannylverbindungen nur, wenn es unbedingt notwendig ist. Die Transferneigung der organischen Reste sinkt mit Steigendem s-Charakter. sp³-hybridisierte Reste werden oft als "Dummyreste" bezeichnet, da sie in Anwesenheit von Resten mit höherem s-Charakter nicht übertragen werden.
Die am Paladium gebundenen Liganden haben ebenfalls Einfluss auf die Reaktion. Je schwächer ein Ligand an das Paladium koordiniert, desto schneller verläuft die Reaktion.
Inhaltsverzeichnis
Reaktionsmechanismus
Der Reaktionsmechanismus der Stille-Kupplung ist weitgehend aufgeklärt.[5]In einem ersten Schritt erfolgt die Reduktion des Palladiumkatalysators (1) zur aktiven Pd(0)-Spezies (2). Die Oxidative Addition des Organohalogenids (3) ergibt das cis-Intermediat welches rasch zum trans-Intermediat 4 isomerisiert.[6] Transmetallierung mit dem Organostannan (5) formt das Intermediat 7, welches nach reduktiver Eliminierung das Produkt (8) ergibt und die aktive Pd(0)-Spezies (2) regeneriert. Die oxidative Addition und die reduktive Eliminierung bewahren die stereochemische Konfiguration der Reaktanten.
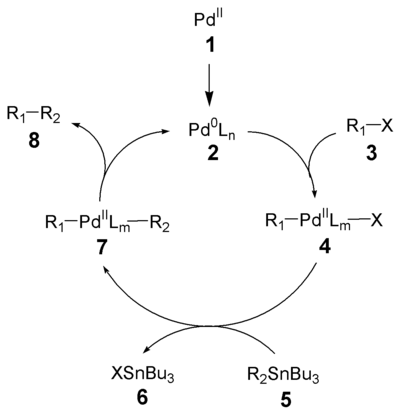
Rate des Ligandentransfers (Transmetallierung) des Zinns:
alkinyl > alkenyl > aryl > allyl = benzyl > α-alkoxyalkyl > alkyl
Variationen
Um die Ausbeute der Reaktion zu erhöhen, setzt man der Reaktionsmischung oft Lithiumchlorid zu. Dieses stabilisiert das in der oxidativen Addition gebildete Intermediat und beschleunigt so die Reaktion.
Reaktivität und Spezifität der Stille-Kupplung kann durch die Zugabe stöchiometrischer Mengen an Kupfer(I)- oder Mn(III)-Salzen erhöht werden. [7][8][9]
In Gegenwart der Cu(I)-Salze zeigt Palladium auf Kohle (Pd/C) eine hohe katalytische Aktivität.[10]
Kritik
Die Atomökonomie der Stille-Kupplung ist gering, was die mögliche technische Nutzung – von Ausnahmen abgesehen – unattraktiv macht.
Siehe auch
- Heck-Reaktion
- Hiyama-Kupplung
- Suzuki-Kupplung
Literatur
- Renaldo, A. F.; Labadie, J. W.; Stille, J. K. Organic Syntheses, Coll. Vol. 8, p.268 (1993); Vol. 67, p.86 (1989). (Volltext)
- Stille, J. K. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524; doi:10.1002/anie.198605081.
- Farina, V.; Krishnamurthy, V.; Scott, W. J. Org. React. 1998, 50, 1–652. (Übersichtsartikel)
- Mitchell, T. N. Synthesis 1992, 803-815; doi:10.1055/s-1992-26230.
Weblinks
- Stille reaction handout der Myers group. (PDF-Datei; 397 kB)
- Stille reaction auf organic-chemistry.org
Einzelnachweise
- ↑ Kosugi, M. et al. Chem. Letters 1977, 301.
- ↑ Milstein, D.; Stille, J. K. J. Am. Chem. Soc. 1978, 100, 3636; doi:10.1021/ja00479a077.
- ↑ Scott, W. J.; Crisp, G. T.; Stille, J. K. Organic Syntheses, Coll. Vol. 8, p.97 (1993); Vol. 68, p.116 (1990). (Article)
- ↑ Stille, J. K.; Echavarren, A. M.; Williams, R. M.; Hendrix, J. A. Organic Syntheses, Coll. Vol. 9, p.553 (1998); Vol. 71, p.97 (1993).
- ↑ Casado, A. L.; Espinet, P. J. Am. Chem. Soc. 1998, 120, 8978–8985; doi:10.1021/ja9742388.
- ↑ Casado, A. L.; Espinet, P. Organometallics 1998, 17, 954–959.
- ↑ Liebeskind, L. S.; Fengl, R. W. J. Org. Chem. 1990, 55, 5359.
- ↑ Farina, V.; Kapadia, S.; Krishnan, B.; Wang, C.; Liebeskind, L. S. J. Org. Chem. 1994, 59, 5905.
- ↑ Liebeskind, L. S.; Peña-Cabrera, E. Organic Syntheses, Coll. Vol. 10, p.9 (2004); Vol. 77, p.135 (2000). (Article)
- ↑ Roth, G. P.; Farina, V.; Liebeskind, L. S.; Peña-Cabrera, E. Tetrahedron Letters 1995, 36, 2191.
Wikimedia Foundation.