- Brugada-Syndrom
-
Klassifikation nach ICD-10 I42.88 Sonstige Kardiomyopathien ICD-10 online (WHO-Version 2011) Das Brugada-Syndrom oder Brugada-Brugada-Syndrom ist eine seltene und meist zwar autosomal-dominant, aber typischerweise mit unvollständiger Penetranz vererbte Krankheit des Herzens. Es wird den „Primären angeborenen Kardiomyopathien“ und dort den sog. Ionenkanalerkrankungen zugerechnet. Patienten mit dieser Erkrankung sind scheinbar völlig herzgesund, können aber bereits im Jugend- und frühen Erwachsenenalter einen plötzlichen Herztod erleiden. Eine wirksame Therapie scheint die Implantation eines automatischen Defibrillators (ICD) bei besonders gefährdeten Patienten zu sein; allerdings wurden auch schon mit Ablationstherapie Patienten wirkungsvoll behandelt.
Inhaltsverzeichnis
Geschichte
Als vermutlich eigenständige Erkrankung wurde das Brugada-Syndrom erst in den 90er-Jahren des 20. Jahrhunderts identifiziert.
1989 beschrieben Kardiologen aus Padua sechs Patienten im Alter zwischen 14 und 35 Jahren, die im Zeitraum von 1977 bis 1988 einen unerklärten Herzstillstand durch Kammerflimmern erlitten hatten und erfolgreich wiederbelebt worden waren. Alle sechs Patienten waren vermeintlich herzgesund, drei von ihnen zeigten im Elektrokardiogramm (EKG) auffällige Hebungen der ST-Strecke in den Ableitungen V1-2.[1]
Namensgeber des Syndroms waren die seinerzeit in Belgien tätigen Brüder Pedro und Josep Brugada, die 1992 acht Patienten beschrieben, welche ebenfalls nach einem Herzstillstand erfolgreich wiederbelebt worden waren. Sie alle zeigten im EKG eine besondere Form des Rechtsschenkelblocks, wiesen aber ansonsten keinerlei Zeichen einer organischen Herzkrankheit auf[2]. Die Ursache des Syndroms war seinerzeit noch unklar.
In einer 2006 aktualisierten Klassifikation von Kardiomyopathien der American Heart Association (AHA) wird das Brugada-Syndrom ebenso wie das QT-Syndrom den „Angeborenen primären Kardiomyopathien“ zugeordnet. [3]
Epidemiologie
Die Prävalenz des Brugada-Syndroms ist nicht bekannt, die Inzidenz wird mit fünf bis 66 pro 10.000 Einwohner angegeben. In Südostasien, wo es häufiger zu sein scheint als in Europa und Nordamerika, tritt das Syndrom bei Männern acht mal häufiger auf als bei Frauen. Herzrhythmusstörungen treten dort durchschnittlich im 40. Lebensjahr auf, wobei die Bandbreite vom ersten bis zum 77. Lebensjahr reicht.[4]
Pathogenese und Pathophysiologie
Mittlerweile wird das Brugada-Syndrom zusammen mit einigen anderen seltenen genetisch determinierten Herzkrankheiten wie dem QT-Syndrom und dem familiären Sinusknoten-Syndrom zu den Ionenkanalerkrankungen (engl. channelopathies) gezählt[5]. Ihnen gemeinsam ist eine genetisch bedingte Veränderung der Eiweißmoleküle (Proteine), die den Ionentransport durch die Zellmembranen des Herzmuskels regulieren. Sie führen zu einem gesteigerten (gain-of-function) oder verminderten (loss-of-function) Transport hauptsächlich von Natrium- und Kaliumionen und verändern dadurch die elektrischen Eigenschaften der Zellen.
Ein verantwortlicher Gendefekt konnte bisher nur bei einem kleineren Teil der Patienten mit Brugada-Syndrom identifiziert werden: 15 bis 25 Prozent von ihnen zeigen eine Mutation des Gens SCN5A, das auf dem dritten Chromosom (Genlocus 3p21) kodiert ist.
Symptome und Krankheitsverlauf
Die dem Syndrom zu Grunde liegende Repolarisationsstörung der Herzmuskelzellen ist nicht spürbar, so dass ein Teil der Patienten keinerlei Symptome bemerkt. Die Leitsymptome plötzliche Bewusstlosigkeit (Synkope) und Herzstillstand treten erst durch Herzrhythmusstörungen wie polymorphe ventrikuläre Tachykardien oder Kammerflimmern auf, die durch die veränderte Repolarisation begünstigt werden. Einzelfälle mit einem sehr frühen Auftreten von Symptomen schon bei Neugeborenen sind zwar beschrieben, meist jedoch treten die Symptome erstmals im dritten oder vierten Lebensjahrzehnt auf.
Diagnose
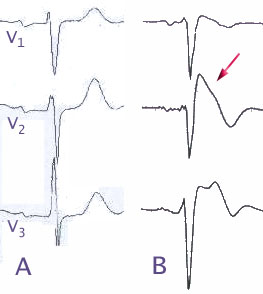
 EKG-Ableitungen V1-3
EKG-Ableitungen V1-3
A Normalbefund
B bei Brugada-Syndrom
Pfeil: zeltförmige ST-HebungEinziges diagnostisches Zeichen für ein Brugada-Syndrom sind typische Veränderung des EKG mit einem Rechtsschenkelblock-ähnlichen Bild, typischerweise etwa einem inkompletten Rechtsschenkelblock, sowie charakteristischen Veränderungen der ST-Strecke in den Ableitungen V1-3.
Diese EKG-Veränderungen können wechselnd ausgeprägt und auch nur zeitweise vorhanden sein. Sie lassen sich durch die Gabe von Medikamenten, die den Natriumkanal blockieren (sog. Natriumkanalblocker) wie Ajmalin, Flecainid oder Procainamid verstärken oder demaskieren. Diese Medikamente werden den Antiarrhythmika der Klasse I nach Vaughan/Williams zugeordnet.
Die EKG-Veränderungen werden in drei verschiedene Typen unterteilt (vgl. Tabelle), die eine unterschiedliche Spezifität für die Diagnose eines Brugada-Syndroms aufweisen.
EKG-Veränderungen beim Brugada-Syndrom „Typ 1“ „Typ 2“ „Typ 3“ J-Amplitude ≥ 0,2 mV ≥ 0,2 mV ≥ 0,2 mV T-Welle negativ positiv oder biphasisch positiv ST-T-Konfiguration gewölbt („coved“) sattelförmig („saddleback“) sattelförmig („saddleback“) terminale ST-Strecke leicht abfallend gehoben ≥ 0,1 mV gehoben < 0,1 mV Da die EKG-Veränderungen bei einigen Patienten gar nicht und bei anderen nur zeitweilig zu beobachten sind, ist die Diagnose des Brugada-Syndroms schwierig und uneinheitlich. Im Jahr 2000 hat eine Arbeitsgruppe der European Society of Cardiology die folgenden Kriterien vorgeschlagen[4]:
- Die Kombination aus einer gewölbten („coved“) ST-Streckenhebung („Typ 1“) in mehr als einer rechtspräkordialen Brustwandableitung (V1-3) und eines der folgenden Ereignisse macht ein Brugada-Syndrom wahrscheinlich: dokumentiertes Kammerflimmern, polymorphe ventrikuläre Tachykardie, plötzlicher Herztod bei Blutsverwandten im Alter von weniger als 45 Jahren, Synkope, Auslösbarkeit bei der elektrophysiologischen Untersuchung oder gewölbte ST-Streckenhebung bei einem Blutsverwandten. Die EKG-Veränderungen können unter Einfluss eines Natriumkanalblockers oder ohne einen solchen aufgetreten sein, andere mögliche Ursachen der Veränderungen sollten aber ausgeschlossen sein.
- ST-Streckenhebung vom „Typ 2“ oder „Typ 3“, die sich nach Gabe eines Natriumkanalblockers zu einer „Typ 1“-Hebung wandeln, sind wie eine solche zu werten. Eine medikamentös verursachte Hebung der ST-Strecke von mindestens 0,2 Millivolt (mV) lässt ein Brugada-Syndrom möglich erscheinen, wenn eines der o. g. klinischen Kriterien vorliegt. Bei Patienten ohne Veränderungen der ST-Strecke nach Gabe eines Natriumkanalblockers ist ein Brugada-Syndrom sehr unwahrscheinlich, ST-Veränderungen von weniger als 0,2 mV gelten als unsicher.
Differentialdiagnostisch ist die beim Brugada-Syndrom als "J-Wave" bezeichnete ST-Hebung von einer ähnlich - wenn nicht i.S. eines „phenotype overlap“ [6] gleich - aussehenden "Epsilon-Welle" als Hinweis auf eine arrhythmogene rechtsventrikuläre Dysplasie (deutsch: ARCM; englisch: arrhythmogenic right ventricular cardiomyopathy / dysplasia ARVC/D) abzugrenzen, was vor allem dann technische Schwierigkeiten bereiten kann, wenn eine vorliegende ARCM nur segmental ausgebildet ist. Weitere, in Betracht zu ziehende Krankheitsbilder sind die RVOT-VT (right ventricular outflow tract ventricular tachycardia) und das typischerweise durch vagotone Reizung ausgelöste sog. idiopathische Kammerflimmern.
Genotyp-Phänotyp-Relation
Bei circa einem Fünftel der Patienten mit Brugada-Syndrom ist ein Defekt des SCN5A-Gens nachweisbar, die übrigen Patienten können bislang genotypisch nicht zugeordnet werden. Patienten mit SCN5A-Mutation unterscheiden sich phänotypisch durch eine verlängerte PQ-Zeit und ein verlängertes HV-Intervall von denen ohne SCN5A-Mutation. Eine PQ-Zeit von ≥ 210 Millisekunden (ms) und ein HV-Intervall von ≥ 60 ms im EKG weisen auf eine SCN5A-Mutation hin. Die Gabe eines Klasse-I-Antiarrhythmikums führte darüber hinaus bei Trägern der SCN5A-Mutation zu einer signifikant längeren PQ-Zeit und QRS-Dauer. Hinsichtlich demografischer Variablen, Anamnese und Familienanamnese hingegen sind zwischen den Gruppen keine Unterschiede festgestellt worden.[7].
Therapie
Die einzige Maßnahme mit nachgewiesener Wirksamkeit gegen den plötzlichen Herztod ist die Implantation eines automatischen Defibrillators (ICD). Sie ist bei allen Patienten mit bereits dokumentierten schwerwiegenden Rhythmusstörungen sinnvoll. Unklar ist, ob auch bislang asymptomatische Patienten einen ICD erhalten sollten, sobald die typischen EKG-Veränderungen aufgefallen sind.
Betablocker sind bei den meisten Patienten mit Brugada-Syndrom vermutlich kontraindiziert, da bei niedriger Herzfrequenz die Gefahr von Kammerflimmern erhöht wird. Patienten mit rezidivierenden Episoden von Kammerflimmern können gelegentlich mittels sympatomimetischer Medikation (z. B. Orciprenalin-Perfusor) zumindest vorübergehend erfolgreich behandelt werden. Bei einigen Patienten kann durch die aktionspotential-verlängernde Wirkung von Chinidin ein Rückgang der typischen EKG-Veränderungen und möglicherweise eine Verminderung des Risikos für Kammerflimmern bewirkt werden.
Daneben ist darauf hinzuweisen, dass auch in einzelnen Fällen das Veröden (Ablation) von elektrophysiologisch ausgemessenen Trigger-Gebieten im Herzmuskel von Patienten, bei denen ein Brugada-Syndrom bekannt war[8], dazu geführt haben soll, dass diese von diesem Moment an keine gefährlichen Herzrhythmusstörungen mehr erlitten.
Literatur
- Douglas P. Zipes et al. (Hrsg.): Braunwald's Heart Disease: A Textbook of Cardiovascular Medicine. 7. Auflage. W.B. Saunders Company, Philadelphia 2004, ISBN 1-41-600014-3
- Youichi A et al.: Assessment of Markers for Identifying Patients at Risk for Life-Threatening Arrhythmic Events in Brugada Syndrome. J Cardiovasc Electrophysiol (2005) 16 (1): 45-51 Online-Version
Belege
- ↑ Martini B et al.: Ventricular fibrillation without apparent heart disease: description of six cases. Am Heart J (1989) 118:1203-1209. PMID 2589161.
- ↑ Brugada P, Brugada J: Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol (1992) 20(6):1391-6. PMID 1309182.
- ↑ Maron BJ et al: Contemporary Definitions and Classification of the Cardiomyopathies. Circulation (2006) 113:1807-1816. PMID 16567565
- ↑ a b Wilde AA: Proposed diagnostic criteria for the Brugada syndrome: consensus report. Circulation (2002) 106:2514-2519. PMID 12417552.
- ↑ Sarkozy A, Brugada P.: Sudden cardiac death and inherited arrhythmia syndromes. J Cardiovasc Electrophysiol (2005) 16 Suppl 1:S8-20. PMID 16138889.
- ↑ Corrado, Domenico, Basso, Cristina, Buja, Gianfranco, Nava, Andrea and Thiene, Gaetano, Brugada syndrome: relationship to other arrhythmogenic syndromes, in: Brugada Syndrome: From Bench to Bedside, pages 111-118, Blackwell Publishing Professional, 2005.
- ↑ Smits JP et al.: Genotype-phenotype relationship in Brugada syndrome: electrocardiographic features differentiate SCN5A-related patients from non–SCN5A-related patients. J Am Coll Cardiol (2002) 40(2):350-6. PMID 12106943.
- ↑ Hsu, Li-Fern, Cauchemez, Bruno, Sanders, Prashanthan, Hocini, Mélèze, Maury, Philippe, Angel Cabrera, Jose, Extramiana, Fabrice, Scavée, Christophe, Takahashi, Y., Rotter, M., Pasquie, J-L., Victor, J., Garrigue, Stéphane, Clémenty, Jacques and Haïssaguerre, Michel, Potential for ablation therapy in patients with Brugada syndrome, in: Brugada Syndrome: From Bench to Bedside, pages 212-220, Blackwell Publishing Professional, ISBN 1405127783, 2005

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Erbkrankheit
- Krankheitsbild in der Kardiologie
Wikimedia Foundation.