- Sialolipidose
-
Klassifikation nach ICD-10 E75.1 Sonstige Gangliosidosen
- Mukolipidose IVICD-10 online (WHO-Version 2011) Die Sialolipidose, auch als Mukolipidose IV bezeichnet, ist eine äußerst seltene autosomal-rezessiv vererbte neurodegenerative lysosomale Speicherkrankheit.
Inhaltsverzeichnis
Inzidenz
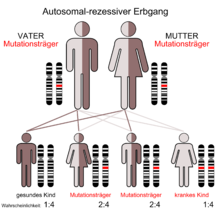 Autosomal-rezessiver Erbgang
Autosomal-rezessiver Erbgang
Die Sialolipidose ist mit einer Inzidenz kleiner als 1 : 1 Million eine ausgesprochen seltene Erkrankung. Bei der Population der Ashkenazi-Juden liegt die Inzidenz mit etwa 1 : 40.000 signifikant höher.[1] Die Heterozygotenfrequenz liegt dort entsprechend bei 1:100.[2][3]
Symptome
Die betroffenen Patienten leiden unter einer psychomotorischen Retardierung und Anomalien der Augen, wie Hornhauttrübung, Netzhautdegeneration oder Schielen (Strabismus).[4] Diese Symptome lassen sich ab dem ersten Lebensjahr, manchmal aber auch später beobachten. Die Progression der Sialolipidose ist in den meisten Fällen eher langsam. In den lysosomalen Einschlußkörpern der Zellen der Erkrankten sind Phospholipide, Ganglioside und Glykosaminoglykane gespeichert.[2]
Genetik und Pathogenese
Der Sialolipidose liegt ein autosomal-rezessiver Erbgang zugrunde. Mutationen im MCOLN1-Gen, das sich auf Chromosom 19 Genlocus p13.3-p13.2[5][6] befindet sind letztlich die Ursache der Erkrankung.[7] MCOLN1 kodiert für das Genprodukt TRPML1 (Mucolipin-1); ein 65 kDa schweres Membranprotein, mit sechs transmembranen Domänen. Es ist aus der Familie der TRP-Kanäle (transient receptor potential). TRPML1 ist ein endo-lysosomaler Kanal für Eisen-Ionen.[8]
Vom MCOLN1-Gen wurden bisher etwa 20 unterschiedliche Mutationen beschrieben, wobei zwei davon 95% aller bei den Aschkenasim gefundenen Mutationen bedingen.[2]
Die Ursache der Akkumulation von Phospholipiden, Ganglioside und Glykosaminoglykane im Lysosom ist wahrscheinlich eine Störung der Endozytose von Komponenten der Zellmembran in die Lysosomen.[2]
Diagnose
Die Diagnose Sialolipidose wird üblicherweise durch den Nachweis der Mutation im MCOLN1-Gen gestellt. Einfacher ist die elektronenmikroskopische Untersuchung der Zellorganellen, die typischerweise bei einer Mucolipidose betroffen sind.[9] Die pränatale Diagnose ist durch elektronenmikroskopische Untersuchung der Amnionzellen oder des Chorions möglich.[10][11]
Eine DNA-Analyse (‚Gentest‘) der Eltern ist ebenfalls vorgeburtlich möglich.[2]
Therapie
Eine Heilung ist bisher ausgeschlossen. Die Behandlung erfolgt rein symptomatisch.[2]
Erstbeschreibung
1974 wurde die Sialolipidose bei erkrankten Ashkenazi-Juden von E. R. Berman und Kollegen erstmals als neue Variante einer Mucolipidose beschrieben.[12]
Weiterführende Literatur
- B. Venugopal u. a.: Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. In: Am J Hum Genet 81, 2007, S. 1070–1083. doi:10.1086/521954 PMID 17924347.
- E. Goldin u. a.: Transfer of a mitochondrial DNA fragment to MCOLN1 causes an inherited case of mucolipidosis IV. In: Hum Mutat 24, 2004, S. 460–465. PMID 15523648
- S. M. Pradhan u. a.: Electronegative electroretinogram in mucolipidosis IV. In: Arch Ophthal 120, 2002, S. 45–50. PMID 11786056
- R. Schiffmann u. a.: Constitutive achlorhydria in mucolipidosis type IV. In: PNAS 95, 1998, S. 1207–1212. PMID 9448310
- K. P. Frei u. a.: Mucolipidosis type IV: characteristic MRI findings. In: Neurology 51, 1998, S. 565–569. PMID 9710036
- R. Bargal und G. Bach u. a.: Mucolipidosis type IV: abnormal transport of lipids to lysosomes. In: J Inherit Metab Dis 20, 1997, S. 625–632. PMID 9323557
- R. D. Folkerth u. a.: Mucolipidosis IV: morphology and histochemistry of an autopsy case. In: J Neuropath Exp Neurol 54, 1995, S. 154–164. PMID 7876885
- D. Chitayat u. a.: Mucolipidosis type IV: clinical manifestations and natural history. In: Am J Med Genet 41, 1991, S. 313–318. PMID 1789285
- B. D. Lake u. a.: A mild variant of mucolipidosis type 4 (ML4). In: Birth Defects Orig Art Ser 18, 1982, S. 391–404. PMID 7171767
- Y. Ben-Yoseph u. a.: Catalytically defective ganglioside neuraminidase in mucolipidosis IV. In: Clin Genet 21, 1982, S. 374–381. PMID 6813002
- B. F. Crandall u. a.: Mucolipidosis IV. In: Am J Med Genet 12, 1982, S. 301–308. PMID 7114093
- L. Caimi u. a.: Mucolipidosis IV, a sialolipidosis due to ganglioside sialidase deficiency. In: J Inherit Metab Dis 5, 1982, S. 218–224. PMID 6820444
- F. Goutieres u. a.: Mucolipidosis IV. In: Neuropaediatrie 10, 1979, S. 321–330.
- S. Merin u. a.: Mucolipidosis IV: ocular, systemic, and ultrastructural findings. In: Invest Ophthal 14, 1975, S. 437–448. PMID 166049
- F. W. Newell u. a.: A new mucolipidosis with psychomotor retardation, corneal clouding, and retinal degeneration. In: Am J Ophthal 80, 1975, S. 440–449. PMID 169696
Weblinks
Einzelnachweise
- ↑ H. Denk, H. P. Dienes und J. Düllmann: Pathologie der Leber und Gallenwege. Verlag Springer, ISBN 3-540-65501-8, S. 255.
- ↑ a b c d e f Sialolipidose bei Orphanet (Datenbank für seltene Krankheiten)
- ↑ A. Raas-Rothschild u. a.: Mucolipidosis type IV: the origin of the disease in the Ashkenazi Jewish population. In: Europ J Hum Genet 7, 1999, S. 496–498. PMID 10352940
- ↑ K. G. Riedel u. a.: Ocular abnormalities in mucolipidosis IV. In: Am J Ophthal 99, 1985, S. 125–136. PMID 3918453
- ↑ S. A. Slaugenhaupt u. a.: Mapping of the mucolipidosis type IV gene to chromosome 19p and definition of founder haplotypes. In: Am J Hum Genet 65, 1999, S. 773–778. PMID 10441585
- ↑ R. Bargal u. a.: Identification of the gene causing mucolipidosis type IV. In: Nature Genet 26, 2000, S. 118–121. PMID 10973263
- ↑ M. Sun u. a.: Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. In: Hum Molec Genet 9, 2000, S. 2471–2478. PMID 11030752
- ↑ X. Dong u. a.: The Type IV Mucolipidosis-Associated Protein TRPML1 is an Endo-lysosomal Iron Release Channel. In: Nature 455, 2008, S. 992. doi:10.1038/nature07311 PMID 18794901
- ↑ N. Amir u. a.: Mucolipidosis type IV: clinical spectrum and natural history. In: Pediatrics 79, 1987, S. 953–959. PMID 2438637
- ↑ G. Kohn u. a.: Prenatal diagnosis of mucolipidosis IV by electron microscopy. In: J Pediat 90, 1977, S. 62–66. PMID 830895
- ↑ A. Ornoy u. a.: Early prenatal diagnosis of mucolipidosis IV. (Letter) In: Am J Med Genet 27, 1987, S. 983–985. PMID 3425607
- ↑ E. R. Berman u. a.: Congenital corneal clouding with abnormal systemic storage bodies: a new variant of mucolipidosis. In: J Pediat 84, 1974, S. 519–526. PMID 4365943

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.