- Glutenzym
-
Strukturformel 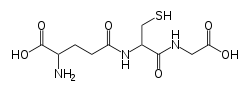
Allgemeines Name Glutathion Andere Namen 2-Amino-5-[1-(carboxymethylcarbamoyl)-2-mercapto- ethyl]amino-5-oxo-pentansäure
Summenformel C10H17N3O6S CAS-Nummer 70-18-8 PubChem 745 Kurzbeschreibung weißer Feststoff[1] Eigenschaften Molare Masse 307,33 g/mol Aggregatzustand fest
Schmelzpunkt Löslichkeit mäßig in Wasser (100 g/l bei 20 °C), fast unlöslich in Ethanol[1]
Sicherheitshinweise Gefahrstoffkennzeichnung [1] keine Gefahrensymbole R- und S-Sätze R: keine R-Sätze S: keine S-Sätze LD50 5000 mg/kg (Maus, oral)[2]
WGK 1[1] Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Glutathion (GSH), auch γ-L-Glutamyl-L-cysteinylglycin, ist ein Peptid, das aus den drei Aminosäuren Glutaminsäure, Cystein und Glycin gebildet wird. Es ist in fast allen Zellen in hoher Konzentration enthalten und gehört zu den wichtigsten als Antioxidans wirkenden Stoffen im Körper. Gleichzeitig ist es eine Reserve für Cystein.
Inhaltsverzeichnis
Biosynthese
Glutathion kann vom Körper aus den Aminosäuren L-Glutaminsäure, L-Cystein und Glycin in einem zweistufigen Prozess synthetisiert werden.
- Unter ATP-Verbrauch wird aus Glutaminsäure und Cystein γ-Glutamylcystein gebildet. Dabei wird eine Isopeptidbindung zwischen der γ-Carboxylgruppe des Glutaminsäurerestes mit der Aminogruppe des Cysteinrestes gebildet. Das daran beteiligte Enzym heißt Glutamatcysteinligase (GCL, auch γ-Glutamylcysteinsynthetase).
- Mit Hilfe der Glutathionsynthase wird unter ATP-Verbrauch Glycin an das terminale Kohlenstoffatom addiert.
Alle Zellen des menschlichen Körpers besitzen die Fähigkeit, GSH zu synthetisieren. Dabei ist die Biosynthese des Stoffs in der Leber essentiell: Mäuse mit gestörter Glutathionproduktion in der Leber sterben innerhalb eines Monats nach der Geburt.[3]
Der Biosyntheseweg findet sich in einigen Bakterien, wie z. B. Cyanobakterien und Proteobakterien, fehlt jedoch in vielen anderen Organismen. Die meisten Eukaryoten sind zur GSH-Synthese fähig, aber etwa Entamoeba und Giardien nicht. Unter den Archaeen können nur Halobakterien selbst synthetisieren.
Funktion
Cystein-Reserve
 Kalottenmodell von Glutathion.
Kalottenmodell von Glutathion.Obwohl GSH als Hauptstoff des reduktiven Pools am bekanntesten ist, entwickelte und stabilisierte sich die Enzym-Maschinerie für die GSH-Synthese wahrscheinlich, weil GSH eine Notreserve für die Aminosäure Cystein darstellt. Eine konstante Versorgung mit Cystein ist unentbehrlich für die Proteinsynthese, aber Cystein ist reaktionsfreudig und geht ständig irreversibel in aerober Umgebung durch Oxidation zu Cysteinsulfinsäure und -sulfonsäure verloren. Außerdem wird es zur Taurinsynthese verwendet. Auf GSH als Kurzzeitspeicher für Cystein kann der Körper daher nicht verzichten. Im menschlichen Blutplasma sind etwa drei Gramm Cystein in Form von GSH enthalten, was einer Reserve für drei Tage entspricht.[4]
Redox-Puffer
GSH kann helfen, zelluläre Makromoleküle, wie etwa Proteine und Membranlipide vor „freien Radikalen“ (reaktive Sauerstoffspezies, ROS) zu schützen. Dabei wird Glutathion oxidiert und geht von seiner monomeren Form GSH in ein Dimer GSSG über.
ROS, die im Verlauf der Zellatmung entstehen können, stellen eine erhebliche Gefahr für zahlreiche Zellbestandteile dar. Reduziertes Glutathion (GSH) besitzt eine freie Thiolgruppe und kann so seinerseits Elektronen auf ROS übertragen und sie so unschädlich machen. Jeweils zwei oxidierte Glutathion-Moleküle verbinden sich unter Ausbildung einer Disulfidbrücke zu einem Glutathion-Disulfid (GSSG). Durch das Enzym Glutathion-Reduktase können aus einem GSSG-Dimer unter Verbrauch von NADPH wieder zwei reduzierte GSH hergestellt werden. Das Redoxpotential von GSH beträgt −240 mV [5] und liegt durch die Aktivität der Glutathion-Reduktase zu 90 % reduziert vor.
Biotransformation
GSH spielt eine wichtige Rolle in Phase II der Biotransformation schädlicher Stoffe. Mit GSH konjugierte Stoffe sind gewöhnlich besser wasserlöslich und können über die Niere ausgeschieden werden. Dabei katalysiert die meist im Zytosol lokalisierte Glutathion-S-Transferase die Reaktion von GSH mit elektrophilem Kohlenstoff. Dabei können Halogen-, Sulfat-, Sulfonat-, Phosphat- und Nitro-Gruppen durch Glutathion substituiert werden. Des weiteren kann GSH an aktivierte Doppelbindungen addiert werden und reaktive Epoxidringe öffnen. Die toxifizierende (giftende) Wirkung umfasst die Aktivierung von vicinalen Dihaloalkanen unter Bildung eines hochreaktiven Episulfoniumringes, sowie eine β-Lyase vermittelte Überführung der GSH-Konjugate in der Niere zu reaktiven Verbindungen.
Weitere Funktionen
In Pflanzen, Nematoden, Algen und Pilzen dient das Glutathion auch als Substrat für die Synthese von Phytochelatinen, die wie Metallothioneine eine wichtige Rolle bei der Bindung und von Schwermetallen spielen.
Eine weitere Aufgabe erfüllt Glutathion bei der Synthese bestimmter Leukotriene (Peptidoleukotriene).
Geschichte
Als Hopkins 1921 ein cysteinhaltiges Peptid in Hefe und Tierzellen beschrieb und Glutathion nannte, war man zunächst der Ansicht, es handele sich um γ-Glutamylcystein. Erst Harington und Mead konnten 1935 durch Totalsynthese die später vermutete tatsächliche Struktur bestätigen.[6][7]
Nahrungsergänzungsmittel
Aufgrund seiner antioxidativen Wirkung wird Glutathion als Nahrungsergänzungsmittel verkauft. Der therapeutische Nutzen von über die Nahrung zugeführtem Glutathion ist allerdings sehr fraglich, da das oral aufgenommene und in die Blutbahn resorbierte Tripeptid nicht in die Zellen aufgenommen wird. Es wird vor der Aufnahme in seine Aminosäurebestandteile zerlegt und im Zellinneren wieder resynthetisiert. Zudem hat jede Körperzelle die Fähigkeit, Glutathion herzustellen. In den meisten Körperzellen liegt Glutathion in hohen Konzentrationen vor. [8]
Die Glutathionproduktion in der Leber kann durch Gabe von Acetylcystein (als Cysteindonor) stimuliert werden.
Als umstrittenes „Krebsmittel“ erlangte Glutathion in einem als Mischung mit Anthocyanen unter dem Namen Recancostat comp. verkauften Präparat Mitte der 90er Jahre Berühmtheit. [9]
Einzelnachweise
- ↑ a b c d e Sicherheitsdatenblatt Merck
- ↑ Sicherheitsdatenblatt Carl Roth
- ↑ Chen Y, et al. (2007) Hepatology 45:1118.
- ↑ David Heber, George L. Blackburn, Vay Liang W. Go, John Milner (Hrsg.): Nutritional Oncology. Academic Press, 2006. S. 310. ISBN 0120883937
- ↑ Aslund F., Berndt K. D., Holmgren A.: Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J Biol Chem. 1997;272(49):30780–6, PMID 9388218
- ↑ Hopkins, FG (1921). On an autoxidisable constituent of the cell. Biochem J. 15, 286-305. Online Version beim Verlag
- ↑ Harington CR, Mead TH: Synthesis of glutathione. In: Biochem. J.. 29, Nr. 7, July 1935, S. 1602–11. PMID 16745829
- ↑ Robinson MK, Ahn MS, Rounds JD, Cook JA, Jacobs DO, Wilmore DW. Parenteral glutathione monoester enhances tissue antioxidant stores. JPEN J Parenter Enteral Nutr. 1992 Sep-Oct;16(5):413-8.
- ↑ www.arzneimittel-und-recht.de: LSG Niedersachsen-Bremen, Urteil vom 15.2.2005, Az.: L 4 KR 44/01, hier online
Weblinks


Wikimedia Foundation.