- Horner-Wadsworth-Emmons-Reaktion
-
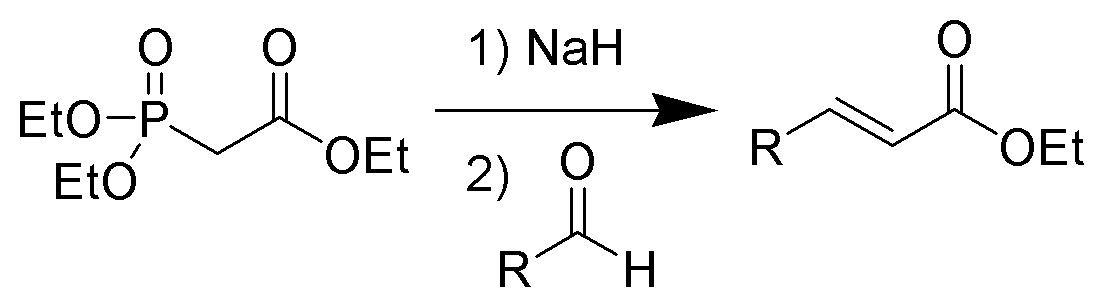
Die Horner-Wadsworth-Emmons Reaktion (kurz: HWE-Reaktion) ist eine chemische Reaktion, mit der stereoselektiv E-Alkene hergestellt werden können. Dazu werden Aldehyde oder Ketone mit den Anionen von organischen Phosphonaten umgesetzt. Die Deprotonierung erfolgt dabei am Kohlenstoff.
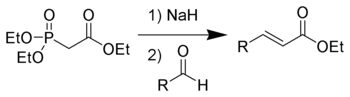
1958 veröffentlichte Leopold Horner eine modifizierte Wittig-Reaktion unter Verwendung phosphonatstabilisierter Carbanionen.[1][2] Wadsworth und Emmons entwickelten diese Reaktion weiter.[3][4]
Im Gegensatz zu den Phosphoryliden der Wittig-Reaktion, sind die phosphonatstabilisierten Carbanionen nucleophiler und basischer. Dementsprechend können diese, anders als die Phosphorylide, alkyliert werden. Die Dialkylphosphatsalze, die als Nebenprodukt anfallen, können einfach durch wässrige Extraktion entfernt werden.
„Übersichtsartikel“: [5][6][7][8]
Inhaltsverzeichnis
Reaktionsmechanismus
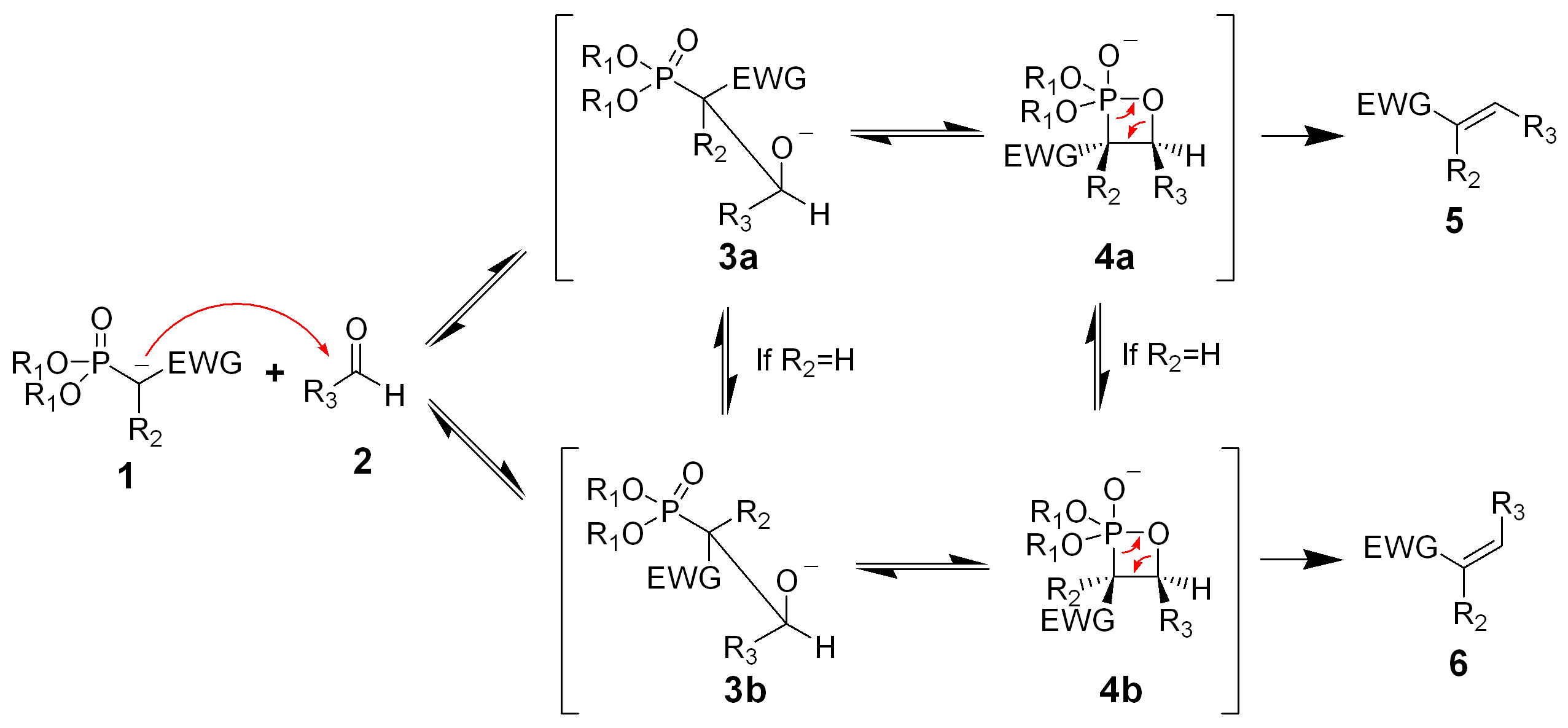
Der Reaktionsmechanismus der Horner-Wadsworth-Emmon-Reaktion ist noch nicht genau bekannt. Sicher beginnt er mit der Deprotonierung des Phosphonats, es bildet sich das Phosphonatcarbanion 1. Man vermutet weiterhin, dass die nucleophile Addition des Carbanions an den Aldehyd 2 (oder auch Keton), die zum Zwischenprodukt 3a oder 3b führt, als der geschwindigkeitsbestimmende Schritt gilt.[9] Wenn R2=H ist, können sich die Intermediate 3a und 4a und die Intermediate 3b und 4b ineinander umwandeln.[10] Eine Eliminierung von 4a und 4b ergibt das (Z)-Alken 5 und das (E)-Alken 6.
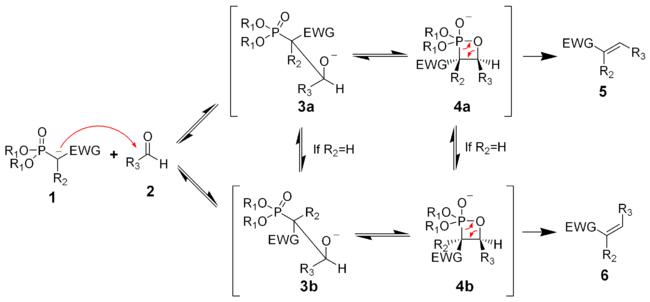
Das Verhältnis der diastereomeren Alkene 5 und 6 ist nicht abhängig vom stereochemischen Ausgang der Carbanionadditon, sondern hängt weitgehend vom Ausmaß des chemischen Gleichgewichts zwischen den Intermediaten ab.
Die Elektronen-ziehende-Gruppe (kurz: EWG – electron withdrawing group) alpha zum Phosphonat ist für die Reaktion unbedingt erforderlich. In Abwesenheit einer „EWG“ ist das Endprodukt der Reaktion das αH-Hydroxyphosphonat 3a bzw. 3b.[11] Diese α-Hydroxyphosphonate können mit Diisopropylcarbodiimiden in Alkene umgewandelt werden.[12]
Stereoselektivität
Die HWE-Reaktion bevorzugt die Bildung von E-Alkenen. Generell gilt, je besser sich das Gleichgewicht zwischen den Intermediaten einstellen kann, desto höher die Selektivität bzw. der E-Alkenanteil.
Disubstituierte Alkene
Thompson und Heathcock veröffentlichten eine systematische Studie der Reaktion von Trimethylphosphonoacetat mit verschiedenen Aldehyden.[13] Während die einzelnen Unterschiede klein waren, zeigte sich ein kumulativer Effekt, der es ermöglicht die Stereoselektivität mit Hilfe der Struktur des Phosphonats zu steuern. Folgende Bedingungen erhöhen die E-Stereoselektivität:
- Ansteigende sterische Hinderung am Aldehyd
- Höhere Reaktionstemperaturen
- Li > Na > K Salze
- Lösungsmittel DME statt THF
In einer weiteren Studie konnte gezeigt werden, dass sterisch anspruchsvolle Phosphonate und „EWGs“ die E-Alkenselektivität ebenfalls erhöhen.
Trisubstitutierte Alkene
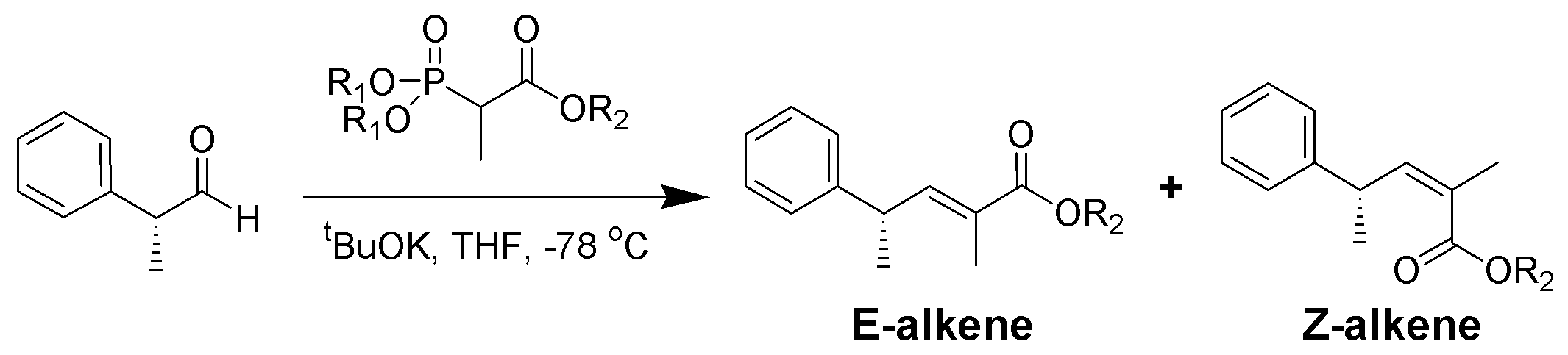
Der sterische Anspruch der Phosphonate und der EWGs zeigt tatsächlich einen Effekt auf die Reaktion von α-verzweigten Phosphonaten mit aliphatischen Aldehyden.[14]
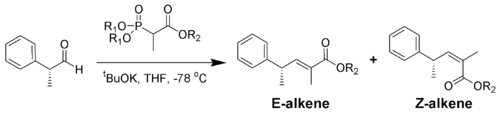
R1 R2 Anteile der Alkene
( E : Z )Methyl Methyl 5 : 95 Methyl Ethyl 10 : 90 Ethyl Ethyl 40 : 60 Isopropyl Ethyl 90 : 10 Isopropyl Isopropyl 95 : 5 Aromatische Aldehyde ergeben meist ausschließlich E-Alkene. Will man Z-Alkene aus aromatischen Aldehyden darstellen, ist die Still-Gennari-Variante (siehe unten) eine Möglichkeit.
Olefinierung von Ketonen
Die Stereoselektivität der HWE ist hier gering bis durchschnittlich.
Variationen
Basen-empfindliche Substrate
Da viele Substrate empfindlich auf Natriumhydrid reagieren, wurde einige Studien zu milderen Basen durchgeführt. Zum einen „Masamune und Roush“ mit Lithiumchlorid und DBU[15] und „Rathke“ mit Lithium oder Magnesiumhalogenen mit Triethylamin[16] und andere Basen[17][18][19].
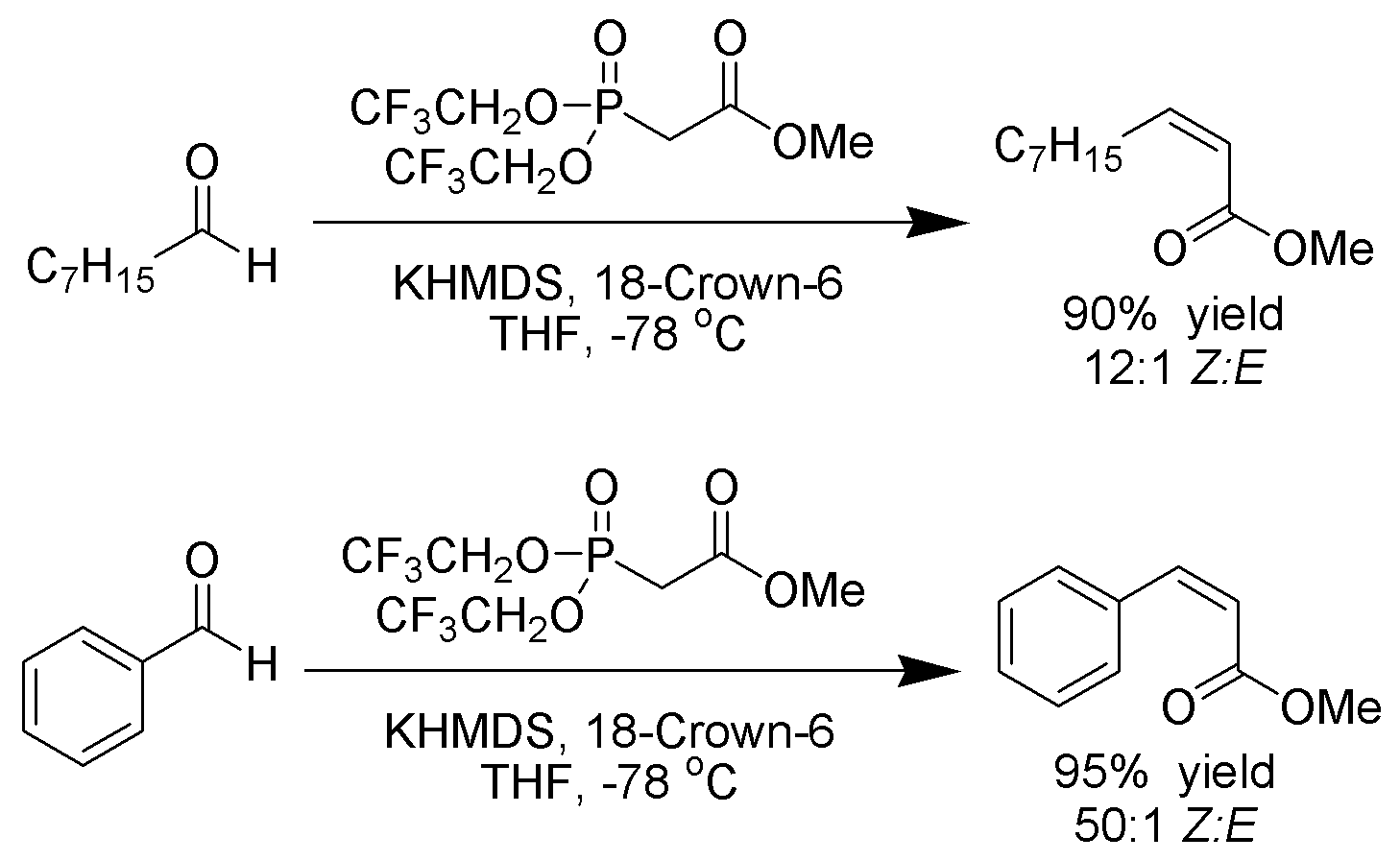
Still-Gennari-Variante
Still und Gennari entwickelten Bedingungen, die eine Reaktion mit sehr hoher Stereoselektivität zu Z-Alkenen ermöglichen.[20] Man nutzt Phosphonate mit elektronenziehenden Gruppen (z. B.: Trifluoroethyl[21]) zusammen mit stark dissozierenden Bedingungen (KHMDS und 18-Kronen-6-ethern in THF).
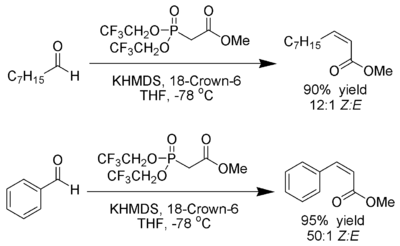
Ando hat hierzu postuliert, dass der Gebrauch elektronenarmer Phosphonate die Eliminierung der Oxophosphonante beschleunigt[22].
Siehe auch
- Michaelis-Arbuzov-Reaktion zur Gewinnung der Reagenzien für die HWE-Reaktion
- Michaelis-Becker-Reaktion
- Peterson-Olefinierung
- Tebbe-Methylenierung
Einzelnachweise
- ↑ Horner, L.; Hoffmann, H.; Wippel, H. G. Chem. Ber. 1958, 91, 61–63
- ↑ Horner, L.; Hoffmann, H.; Wippel, H. G.; Klahre, G. Chem. Ber. 1959, 92, 2499–2505.
- ↑ Wadsworth, W. S., Jr.; Emmons, W. D. Journal of the American Chemical Society 1961, 83, 1733.
- ↑ Wadsworth, W. S., Jr.; Emmons, W. D. Organic Syntheses, Coll. Vol. 5, p.547 (1973); Vol. 45, p.44 (1965) (Article).
- ↑ Wadsworth, W. S., Jr. Org. React. 1977, 25, 73–253.
- ↑ Boutagy, J.; Thomas, R. Chemical Reviews 1974, 74, 87–99.
- ↑ Kelly, S. E. Comp. Org. Syn. 1991, 1, 729–817.
- ↑ Maryanoff, B. E.; Reitz, A. B. Chemical Reviews 1989, 89, 863–927.
- ↑ Larsen, R. O.; Aksnes, G. Phosphorus Sulfur 1983, 15, 218–219.
- ↑ Lefèbvre, G.; Seyden-Penne, J. J. Chem Soc., Chem. Commun. 1970, 1308–1309.
- ↑ Corey, E. J.; Kwiatkowski, G. T. Journal of the American Chemical Society 1966, 88, 5654–5656.
- ↑ Reichwein, J. F.; Pagenkopf, B. L. Journal of the American Chemical Society 2003, 125, 1821–1824.
- ↑ Thompson, S. K.; Heathcock, C. H. J. Org. Chem. 1990, 55, 3386–3388.
- ↑ Nagaoka, H.; Kishi, Y. Tetrahedron 1981, 37, 3873–3888.
- ↑ Blanchette, M. A.; Choy, W.; Davis, J. T.; Essenfeld, A. P.; Masamune, S.; Roush, W. R.; Sakai, T. Tetrahedron Letters 1984, 25, 2183–2186.
- ↑ Rathke, M. W.; Nowak, M. J. Org. Chem. 1985, 50, 2624.
- ↑ Paterson, I.; Yeung, K.-S.; Smaill, J. B. Synlett 1993, 774.
- ↑ Simoni, D.; Rossi, M.; Rondanin, R.; Mazzali, A.; Baruchello, R.; Malagutti, C.; Roberti, M.; Invidiata, F. P. Org. Lett. 2000, 2, 3765–3768.
- ↑ Blasdel, L. K.; Myers, A. G. Org. Lett. 2005, 7, 4281–4283.
- ↑ Still, W. C.; Gennari, C. Tetrahedron Letters 1983, 24, 4405–4408.
- ↑ Patois, C.; Savignac, P.; About-Jaudet, E.; Collignon, N. Organic Syntheses, Coll. Vol. 9, p.88 (1998); Vol. 73, p.152 (1996). (Article).
- ↑ Ando, K. J. Org. Chem. 1997, 62, 1934–1939.
Wikimedia Foundation.