- Hypertrophe Kardiomyopathie
-
Die Hypertrophe Kardiomyopathie (auch: hypertrophische Kardiomyopathie; HCM; früher: idiopathische hypertrophe subaortale Stenose; IHSS) ist eine angeborene Erkrankung und gehört zur großen Gruppe der Kardiomyopathien (gr. kardio (καρδία) Herz, gr. myo (μυς) Muskel, gr. páthos (πάθος) Leiden; Erkrankung der Herzmuskulatur). Sie ist charakterisiert durch eine meist asymmetrische Verdickung (Hypertrophie) der Muskulatur der linken Herzkammer. Bei einem Teil der Fälle kommt es zu einer unter Belastung zunehmenden Verengung (Obstruktion) der linksseitigen Ausflussbahn und im Verlauf zu einer Versteifung des Herzmuskels (Compliancestörung). Hauptbeschwerden sind Luftnot unter Belastung sowie teilweise gefährliche Herzrhythmusstörungen. Behandelt wird die Erkrankung mit herzkraftsenkenden Medikamenten und, im Falle einer Verlegung des Ausflusstraktes, mit einer herzkathetergeführten oder operativen Muskelentfernung.
Klassifikation nach ICD-10 I42.1 Hypertrophische obstruktive Kardiomyopathie I42.2 Hypertrophische nichtobstruktive Kardiomyopathie ICD-10 online (WHO-Version 2011) 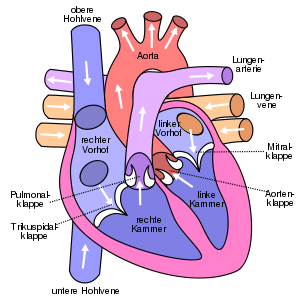 Schema des menschlichen Herzens
Schema des menschlichen Herzens
Inhaltsverzeichnis
Ursache - Ätiologie
Die Erkrankung ist angeboren und tritt familiär gehäuft auf. Sie wird autosomal dominant vererbt. Männer und Frauen sind gleich häufig betroffen. Die Prävalenz liegt bei mindestens 1:500.[1]
Genotyp
Über 200 Gendefekte auf insgesamt zehn Genen, die Proteine des kardialen Sarkomers kodieren, sind bekannt. Die wichtigsten Defekte (über 50 %) liegen im Aufbau der beta-myosin heavy chain (ein Myosinfilament, der erste identifizierte Gendefekt), im myosinbinding protein C und im Troponin-T.[1]
Phänotyp
Die phänotypische Ausprägung ist meist nicht ausschließlich abhängig von einer einzelnen Mutation, sondern wird durch „Modifier-Gene“ und „Umgebungsfaktoren“ mitbedingt. Das klinische Bild (linksventrikuläre Hypertrophie in der Echokardiographie, EKG-Veränderungen) kann, trotz nachgewiesenem Gendefekt, völlig fehlen. Kinder unter 13 Jahren sind üblicherweise ausschließliche „stille Genträger“.[1]
Feingewebliche Veränderungen - Pathologie
Das feingewebliche Bild ist nicht spezifisch. Es findet sich eine Verzweigungsstörung der hypertrophierten Herzmuskelzellen mit Fehlen der normalen parallelen Anordnung durch vermehrte seitliche Verzweigungen, die durch Seit-zu-Seit-Verbindungen ersetzt wurden (myozytäres Disarray). Weiter findet sich ein bindegewebiger (fibrotischer) Umbau des Interstitiums. Die mikroskopischen Veränderungen sind nicht auf die makroskopisch hypertrophierten Bereiche beschränkt. Ein ähnliches Bild, nur bei weitem nicht so ausgeprägt, zeigt sich auch bei anderen mit Herzmuskelhypertrophie einhergehenden Erkrankungen.[2]
Krankheitsentstehung - Pathogenese
Bei etwa einem Viertel der betroffenen Patienten befindet sich die Herzmuskelverdickung im Ausflusstrakt der linken Herzkammer, wodurch es je nach Schweregrad unter Belastung (körperlich oder medikamentös) oder auch in Ruhe zu einer Einengung (Obstruktion) des Ausflusstraktes kommt. Dadurch entsteht eine funktionelle Aortenstenose mit erhöhter Druckbelastung der linken Kammer. Die HCM wird dementsprechend in eine obstruktive (HOCM) und eine nicht-obstruktive Form (HNCM) unterteilt. Bei nur unter Belastung nachweisbarer Obstruktion wird auch von einer „HCM mit dynamischer Obstruktion“ gesprochen. Etwa 12 % der Patienten mit einer HOCM leiden unter einer atypischen Form bei der die Obstruktion nicht subaortal sondern mitventrikulär liegt.[3]
Neben der Einengung der Ausflussbahn bei der HOCM führt die Muskelverdickung bei beiden Formen der HCM zu einer Muskelversteifung. Hierdurch kann sich die Herzkammer in ihrer Erschlaffungsphase (Diastole) nur eingeschränkt füllen, wodurch Blut in die Lungenvenen zurückstaut mit nachfolgender Atemnot. Man spricht von einer diastolischen Herzinsuffizienz (diastolische Compliancestörung). Bei der HOCM nimmt die Muskelversteifung durch die vermehrte Arbeit (Pumpleistung gegen die Enge des Ausflusstraktes) noch zusätzlich zu.
Im Weiteren findet sich bei der HOCM, durch die auftretende Flussbeschleunigung im Bereich der verengten Ausflussbahn, eine Sogwirkung auf die Mitralklappe (Venturi-Effekt), wodurch diese in meist nur mäßiger Ausprägung undicht werden kann (Mitralinsuffizienz).
Hauptproblem neben der Belastungseinschränkung ist die Neigung zu schwerwiegenden, vor allem unter Belastung auftretenden Rhythmusstörungen. Diese können mit Synkopen (kurze Bewusstlosigkeit) und dem plötzlichen Herztod einhergehen. So sind plötzliche Todesfälle von Menschen unter 35 Jahren beim Sport häufig durch eine HCM verursacht. Das jährliche Todesfallrisiko bei Erwachsenen mit HCM ist individuell zu beurteilen und beträgt etwa 1 %, bei Kindern ist es höher.
Klinische Erscheinungen
Die Patienten sind, besonders bei der nicht-obstruktiven Form (HNCM), häufig asymptomatisch. Wenn Symptome auftreten, sind diese meist nicht wegweisend (Luftnot, Angina pectoris, Rhythmusstörungen, Schwindel, Synkopen, plötzliche Todesfälle).
Untersuchungsmethoden - Diagnostik
Die HCM muss von einer reaktiven Herzmuskelhypertrophie durch Sport (Athletenherz) oder langjährigen Bluthochdruck (Hypertonieherz) sowie von einer Erkrankung der Aortenklappe (Aortenklappenstenose) abgegrenzt werden.
Anamnese und körperliche Untersuchung
Neben nur wenig wegweisenden Beschwerdeangaben in der Anamnese (siehe oben) fällt bei der körperlichen Untersuchung im Rahmen der Auskultation ein Systolikum auf, das unter Belastung (z. B. 10 Kniebeugen) oder auch unter einem Pressmanöver (Valsalva-Manöver) zunimmt. Dadurch lässt sich die HOCM relativ sicher von der Aortenklappensteose oder der Mitralklappeninsuffiizenz abgrenzen, da deren Geräusche beim Valsalva-Manöver leiser werden.
Technische Untersuchungsverfahren
Im EKG sind eventuell Zeichen der linksventrikulären Hypertrophie (Sokolow-Lyon-Index), Q-Zacken und Repolarisationsstörungen zu sehen, diese sind allerdings unspezifisch.[2]
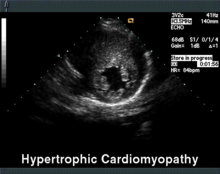 Verdicktes Septum in parasternal kurzer Achse
Verdicktes Septum in parasternal kurzer AchseDie Echokardiographie ist das diagnostische Mittel der Wahl. Neben der Septumhypertrophie kann hier häufig auch ein verlagertes Mitralklappensegel gesehen werden, welches in der Systole ein Bewegung zum Septum macht und den Ausflusstrakt zusätzlich einengt (systolic anterior movement = SAM). Im weiteren kann der Ruhe-Gradient im linksventrikulären Ausflußtrakt (LVOT), d. h. ein Drucksprung zwischen linker Herzkammer und Hauptschlagader, gemessen sowie die Herzmuskelversteifung beurteilt werden.
In der Belastungs-Echokardiographie kann der Gradient im LVOT vor und nach Belastung bestimmt werden, womit ein objektiver Parameter zu Verlaufs- und Therapiekontrolle zur Verfügung steht.
Die Kernspintomographie kann auch atypische (Verteilungs-)Formen zuverlässig darstellen und ist deshalb bei klinischem Verdacht und fehlendem Nachweis der HOCM in der Echokardiographie indiziert. Sie kann fleckförmige Narben im hypertrophierten Myokard nachweisen, die als Risikofaktor für Arrhythmien und plötzlichen Herztod gelten. Auch die Flussbeschleunigung im Ausflusstrakt sowie die narbigen Veränderungen nach therapeutischer Septumembolisation sind kernspintomographisch gut darstellbar.[4]
Die Herzkatheteruntersuchung bietet die Möglichkeit einer direkten Druckmessung im Herzen zur Bestimmung des Gradienten im LVOT und der Bestimmung des Ausmaßes der Herzmuskelversteifung sowie zum Ausschluss anderer Erkrankungen. Wird während der Katheteruntersuchung eine ventrikuläre Extrasystole ausgelöst, kommt es zu einer Vervielfachung des Druckgradienten sowie zu einem fehlenden Anstieg des sytolischen Blutdrucks beim postextrasystolischen Schlag (sog. Brockenbrough-Phänomen). Dies gilt als pathognomisch für die HOCM.
Labortechnisch besteht die Möglichkeit zur Gendiagnostik. Diese ist allerdings sehr teuer und wird daher z. Z. nur im Rahmen von Studien durchgeführt.
Eine weitere sehr einfache Methode zur Verlaufs- und Therapiekontrolle ist der 6-Minuten-Gehtest.
Angehörigenscreening
Verwandte ersten Grades sollten auf das Vorliegen einer HCM untersucht werden. Kinder erkrankter Eltern haben ein 50%iges Risiko, ebenfalls Träger des Gendefektes zu sein. Falls keine Gendiagnostik möglich ist (s. o.), sollten Kinder und Jugendliche zwischen dem 12. und 18. Lebensjahr jährlich und nach dem 18. Lebensjahr alle fünf Jahre echokardiographisch untersucht werden. Vor dem 12. Lebensjahr wird ein Screening nur bei Kindern empfohlen, die aus einer Hochrisikofamilie stammen oder Leistungssport betreiben.[1]
Therapie
Konservative Maßnahmen
Körperliche Belastung in Abhängigkeit von der Ausprägung der Erkrankung ist erlaubt, jedoch sollte kein Leistungssport und keine Sportarten mit plötzlich einsetzenden Maximalbelastungen (Fußballspiel) betrieben werden. Hierbei besteht eine erhöhte Gefahr bösartiger Herzrhythmusstörungen.
Bei beiden Formen der HCM werden Medikamente, welche die Leistung der linken Herzkammer herabsetzen („weniger ist mehr“), gegeben. Hierzu gehören Betablocker oder Calciumantagonisten vom Verapamil-Typ.
Bei schwerwiegenden Rhythmusstörungen kommen Antiarrhythmika zum Einsatz.
Medikamente, die die Kontraktionskraft des Herzmuskels stärken, wie Digitalis oder Katecholamine, verstärken auch die Obstruktion bei der HOCM und dürfen daher nicht verwendet werden. Ebenso führen Vor- oder Nachlast-senkende Medikamente wie Nitroverbindungen, ACE-Hemmer oder AT1-Antagonisten zu einer Zunahme des Gradienten im Ausflusstraktes und sind ebenso kontraindiziert.
Interventionelle Maßnahmen
Bei einer HOCM sollte bei Beschwerden die definitive Behandlung der Obstruktion ab einem Ruhegradienten im LVOT > 30 mmHg oder bei einem Gradienten nach Provokation angestrebt werden.[5] Die Ausbildung einer diastolischen Compliancestörung kann hierdurch vermindert werden. Es stehen drei Therapieoptionen zur Verfügung.
Transkoronare Ablation der Septumhypertrophie (TASH)
Die Transcoronare Ablation der Septumhypertrophie (TASH)[5], alcohol septal ablation (ASA)[1] oder Perkutane transluminale septale Myokard-Ablation (PTSMA) ist das interventionelle Verfahren der Wahl zur Behandlung der Obstruktion einer HOCM.[6] Bei dieser von Sigwart erstmals im Juni 1994 am Royal Brompton Hospital in London durchgeführten Methode[7] wird mittels eines Herzkatheters zunächst der erste Septalast des Ramus interventricularis anterior (RIVA; LAD) aufgesucht und vorübergehend mit einem Ballon verschlossen. Falls es hierauf zu einem Abfall des Gradienten im linksventrikulären Ausflusstrakt kommt, wird reiner Alkohol durch den Ballon in dieses Gefäß gegeben, um so einen umschriebenen Infarkt im Bereich der Obstruktion auszulösen. Dieser Bereich schrumpft und die Obstruktion verringert sich in den folgenden Monaten.
Die Erfolgsquote liegt bei über 88 %, die Letalität unter 1,2 %.[6]. In etwa 10 % der Fälle wird ein permanenter AV-Block III° verursacht, so dass die Notwendigkeit zur Implantation eines Herzschrittmachers besteht. In 15 % der Fälle ist eine zweite TASH notwendig.[8] Ein sehr seltener, potentiell lebensbedrohlicher Begleiteffekt ist das akute coronare No-Flow-Phänomen (ACNF).
Eine bisher nicht etablierte Variante der TASH ist die Ablation des Septalastes mit Hilfe eines Cyanoacryls.[9]
Endokardiale Radiofrequenzablation der Septumhypertrophie (ERASH)
Die herzkathetergeführte Radiofrequenzablation zur Behandlung von Herzrhythmusstörungen gibt es bereits seit geraumer Zeit. Zur Behandlung der HOCM wurde sie erstmals durch Lawrenz 2004[10] eingesetzt. Bisher wird die Methode nur an einzelnen Zentren durchgeführt. Verwendung findet sie nach erfolgloser TASH. Hierbei wird die elektrische Energie mittels Herzkatheter im Obstruktionsareal am rechtsventrikulären Septum abgegeben. Wie bei der TASH wird hierdurch eine Narbe hervorgerufen, um den Gradienten im linksventrikulären Ausflusstrakt zu verringern. Für eine abschließende Beurteilung der langfristigen Wirksamkeit liegen noch nicht ausreichend Daten vor.
Unter dem Begriff der Radiofrequenzkatheterablation (RFKA) wurde die Technik erstmals bei zwei Kindern (5 und 11 Jahre) 2005 erfolgreich angewandt.[11]
Transaortale subvalvuläre Myektomie (TSM)
Die Operation ist heute die Alternative der letzten Wahl. In der seit mehreren Jahrzehnten bekannten Herzoperation nach Morrow wird durch die Aortenklappe hindurch überschüssiges Muskelgewebe im Ausflusstrakt der linken Herzkammer entfernt. Erfolgsquote und Risiken sind ähnlich der TASH, das Verfahren ist allerdings deutlich invasiver.
Supportive Maßnahmen
Zum Schutz vor bösartigen Herzrhythmusstörungen wird in Abhängigkeit von der Familienanamnese (plötzlicher Herztod bei Verwandten ersten Grades) die primärprophylaktische Implantation eines Defibrillators (ICD) empfohlen. Sind bösartige Herzrhythmusstörungen bereits dokumentiert, sollte in jedem Fall ein ICD implantiert werden.
Eine alleinige Zwei-Kammer-Schrittmachertherapie mit einer verkürzten Vorhof-Kammer-Überleitungszeit hat sich als nicht ausreichend wirksam herausgestellt. Eine Verminderung des Gradienten im LVOT kann zwar nachgewiesen werden, die subjektive Besserung des Befindens liegt aber im Bereich des Placebo-Effekt.[5] Unterstützend kann die Implantation bei Patienten empfohlen werden, die zusätzlich unter behandlungsbedürftigen tachykarden oder bradykarden Herzrhythmusstörungen leiden oder die einer invasiven Therapie (s. o.) ablehnend gegenüberstehen.[12]
Prognose
Die HCM ist eine bisher nicht heilbare, jedoch bei frühzeitiger Diagnose häufig gut behandelbare Erkrankung. Insgesamt ist die HOCM die meist schwerwiegendere Form. Das relative Risiko für Komplikationen wie Herzinsuffizienz im Stadium NYHA III-IV, Tod durch Schlaganfall oder Herzschwäche ist 2,0. D. h. das Risiko ist doppelt so hoch bei der HOCM im Vergleich zur HNCM.[1]
Geschichte
Die Erkrankung wurde 1869 von Liouville und Hallopeau zuerst beschrieben. Seit ihrer Beschreibung im Jahr 1957 durch Sir Russell Brock ist sie allgemein als klinische Entität akzeptiert.
Literatur und Quellen
- Christian Mewis, Reimer Rissen, Ioakim Spyridopoulos (Hrsg.) (Hrsg.): Kardiologie compact. Alles für Station und Facharztprüfung. 2. unveränderte Auflage. Georg Thieme, Stuttgart u. a. 2006, ISBN 3-13-130742-0.
- Gerd Herold: Innere Medizin. Eine vorlesungsorientierte Darstellung. Unter Berücksichtigung des Gegenstandskataloges für die Ärztliche Prüfung. Mit ICD 10-Schlüssel im Text und Stichwortverzeichnis. Herold, Köln 2007.
- Prinz, C et al.: Diagnostik und Therapie bei hypertropher Kardiomyopathie. In: Dtsch Arztebl Int. Nr. 108(13), 2011, S. 209-215 (Abstract).
- Einzelnachweise
- ↑ a b c d e f American College of Cardiology / European Society of Cardiology Clinical Expert; Consensus Document on Hypertrophic Cardiomyopathy; Journal of the American College of Cardiology; Vol. 42, No. 9, 2003; ISSN 0735-1097
- ↑ a b Hengstenberg, C.; Genetik der familiären hypertrophischen Kardiomyopathie; Dtsch Arztebl 1996; 93(9): A-532 / B-430 / C-406; Artikel
- ↑ Kuhn H., Mercier J., Köhler E., Frenzel H., Hort W., Loogen F.; Differential diagnosis of hypertrophic cardiomyopathies: typical (subaortic) hypertrophic obstructive cardiomyopathy, atypical (midventricular) hypertrophic obstructive cardiomyopathy and hypertrophic non obstructive cardiomyopathy; Eur Heart J 1983; 4(Suppl F): 93-104
- ↑ Michael Schäfers et al: Nichtinvasive kardiale Bildgebung. ecomed Medizin, 2008, ISBN 978-3-609-16282-9.
- ↑ a b c Mewis, Riessen, Spyridopoulos (Hrsg.): Kardiologie compact - Alles für Station und Facharztprüfung. 2 Auflage. Thieme, Stuttgart, New York 2006, ISBN 3-13-130742-0, S. 396+397.
- ↑ a b Faber L, Seggewiss H, Gietzen FH, Kuhn H, Boekstegers P, Neuhaus L, Seipel L, Horstkotte D.; Catheter-based septal ablation for symptomatic hypertrophic obstructive cardiomyopathy: follow-up results of the TASH-registry of the German Cardiac Society; Z Kardiol. 2004 Jan;93(1):23-31; PMID 16049653, dt. Abstract
- ↑ Sigwart U.; Non-surgical myocardial reduction for hypertrophic obstructive cardiomyopathy; Lancet. 1995 Jul 22;346(8969):211-4; PMID:7616800
- ↑ Kuhn H.; Transcoronary ablation of septal hypertrophy (TASH): a 5-year experience; Z Kardiol. 2000 Jun;89(6):559-564; PMID 10929441
- ↑ Oto A, Aytemir K, Deniz A.; New approach to septal ablation: glue (cyanoacrylate) septal ablation; Catheter Cardiovasc Interv. 2007 Jun 1;69(7):1021-5; PMID 17525960
- ↑ Lawrenz T., Kuhn H.; Endocardial radiofrequency ablation of septal hypertrophy. A new catheter-based modality of gradient reduction in hypertrophic obstructive cardiomyopathy; Z Kardiol. 2004 Jun;93(6):493-9; PMID 15252744; dt. Abstract
- ↑ M. Emmel, N. Sreeram, J. V. deGiovanni, K. Brockmeier; Radiofrequency catheter septal ablation for HOCM in childhood; Kardiol 94:699-703 (2005); PMID 16598534; dt. Abstract
- ↑ Strunk-Mueller C., Gietzen F., Kuhn H.; Schrittmachertherapie der hypertrophisch obstruktiven Kardiomyopathie; Herzschrittmacherther Elektrophysiol. 2004 Jul;15(1):i47-i53; dt. Abstract
Weblinks
- Updated Meta-Analysis of Septal Alcohol Ablation Versus Myectomy for Hypertrophic Cardiomyopathy, 2010, doi:10.1016/j.jacc.2009.09.047
- Cardiomyopathy, familial hypertrophic; CMH bei Online Mendelian Inheritance in Man.
- Kuhn, H. (2009): How to perform a safe and effective TASH (englisch). Abgerufen am 27. Oktober 2009..
- Raute-Kreinsen, U.: Morphologie von Nekrosen und Reparation nach alkoholinduzierter transcoronarer Ablation der Septumhypertrophie bei HOCM. In: Elsevier Verlag (Hrsg.): Pathology - Research & Practice. 2007-04-19..

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorie:- Krankheitsbild in der Kardiologie

Wikimedia Foundation.