- Arrhythmogene rechtsventrikuläre Kardiomyopathie
-
Klassifikation nach ICD-10 I42.- Kardiomyopathie ICD-10 online (WHO-Version 2006) Als Kardiomyopathien werden Erkrankungen des Herzmuskels bezeichnet.
Die Kardiomyopathien stellen eine Gruppe von Erkrankungen dar, deren wesentliches Merkmal die direkte Beteiligung des Herzmuskels selbst ist. Sie sind nicht die Folge von Erkrankungen des Herzbeutels (Perikard), eines Bluthochdrucks, angeborener Herzfehler oder einer Herzklappenerkrankung.
Es existieren unterschiedliche und umstrittene Klassifikationen der Kardiomyopathien.
Die zur Zeit noch geläufigste ist die 1980 durch die WHO erstellte und 1995 wesentlich erweiterte Klassifikation: „Herzmuskelerkrankungen, die mit einer Fehlfunktion des Herzens einhergehen“.
Aufgrund zahlreicher neuer Erkenntnisse wurde im März 2006 von der Amerikanischen Herzgesellschaft (American Heart Association, AHA) eine aktualisierte Definition und Klassifikation der Kardiomyopathien [1] vorgeschlagen. In dieser neuen Klassifikation werden primäre von sekundären Kardiomyopathien unterschieden. Die primären K. wiederum werden in angeborene, erworbene und Mischformen unterteilt.
- „Kardiomyopathien sind eine heterogene Gruppe von Krankheiten des Herzmuskels, die mit mechanischen und/oder elektrischen Funktionsstörungen einhergehen und üblicherweise (aber nicht zwingend) eine unangemessene Hypertrophie oder Dilatation der Herzkammern verursachen. Ihre Ursachen sind vielfältig und häufig genetisch bedingt. Kardiomyopathien begrenzen sich entweder auf das Herz oder sind Teil einer allgemeinen Systemerkrankung, führen oft zu kardiovaskulär bedingten Todesfällen oder einer fortschreitenden Behinderung durch Herzversagen.“
Geschichte
In der Mitte des 18. Jahrhunderts war allein die chronische Myokarditis als Herzmuskelerkrankung bekannt. Um 1900 wurde der Begriff der primären Herzmuskelerkrankung geprägt, und erst 1957 kam der Begriff der Kardiomyopathie auf. Bis 1980 gab es mehrere Definitionen, als die WHO die Kardiomyopathie als "Herzmuskelerkrankung unbekannter Ursache“ bezeichnete. Die WHO-Klassifikation von 1995 erweiterte den Begriff auf „Herzmuskelerkrankungen, die zu Fehlfunktionen des Herzens führen“. Neue Erkrankungen wie die arrhythmogene rechtsventrikuläre und die restriktive Kardiomyopathie wurden eingeschlossen.
Primäre Kardiomyopathien
Angeborene primäre Kardiomyopathien
Hypertrophe Kardiomyopathie
siehe auch Hauptartikel hypertrophe Kardiomyopathie
Die Hypertrophische Kardiomyopathie (HCM) ist durch eine meist asymmetrische Verdickung der Muskulatur der linken Herzkammer charakterisiert.Arrhythmogene rechtsventrikuläre Kardiomyopathie
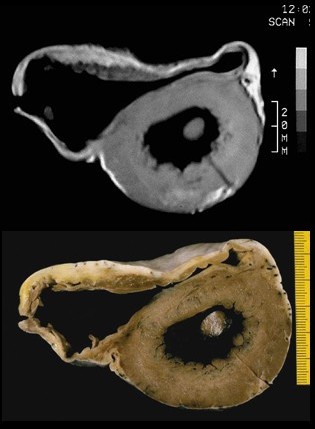
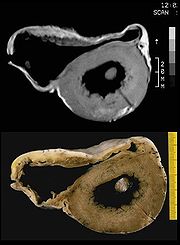 CT-Aufnahme und pathologisches Präparat einer ARVCM
CT-Aufnahme und pathologisches Präparat einer ARVCMArrhythmogene rechtsventrikuläre Kardiomyopathie (ARVCM), früher auch arrhythmogene rechtsventrikuläre Dysplasie genannt.[2] Überwiegend angeborene Erkrankung, die in Norditalien (Venetien) häufiger vorkommt, aber auch in Deutschland gefunden wird. Es findet sich ein zunehmender Ersatz der Muskulatur der rechten Herzkammer durch Fettgewebe, hierdurch vergrößert sich die rechte Herzkammer. Selten finden sich Einschränkungen der Pumpfunktion, aber häufig ein bei körperlicher Belastung wie Sport ausgelöster plötzlicher Herztod (PHT) oder „Beinahe“-PHT, insbesondere bei jungen Menschen. Diagnose: Echokardiografie, MRT, EKG, McKenna-Score. Therapie: Implantation eines ICD (implantierbarer Cardioverter/Defibrillator), keine sportliche Belastung, ggf. Herztransplantation, Untersuchung von Familienangehörigen von Betroffenen. Prophylaxe (Vorbeugung) in Italien und USA: Untersuchung aller Mitglieder von Sportvereinen. Der spanische Fußballspieler Antonio Puerta litt an arrhythmogener rechtsventrikulärer Kardiomyopathie. Er verstarb drei Tage nachdem er während eines Ligaspiels im August 2007 mehrere Herzstillstände erlitt.
Linksventrikuläre Hypertrabekulation
Erst vor wenigen Jahren entdeckte, angeborene Herzmuskelerkrankung mit schwammartig aufgetriebener Muskulatur vor allem in der Spitze der linken Herzkammer, die tiefe Aushöhlungen (Sinusoide) zwischen Muskelfasern (Trabekeln) aufweist, die mit der Herzhöhle verbunden sind. Bei der „Isolierten Nonkompaktion des Herzmuskels“ (Syn: Non-Compaction-Kardiomyopathie, linksventrikulären Hypertrabekulation, spongy myocardium) hat sich der Herzmuskel aus seinem losen Maschennetz während der frühen Embryonalphase nicht weiter verdichtet (schwammiges Myokard). Gehäuft bei Skelettmuskelerkrankungen, auch in Kombination mit komplexen zyanotischen Herzfehlern. Selten.
Die Diagnose wird durch Echokardiografie, MRT oder Angiografie der linken Herzkammer bei einer Herzkatheteruntersuchung gestellt. Unklar ist der klinische Verlauf. Fälle von schwerem Herzversagen, Thromboembolie, Arrhythmien und plötzlichem Herztod sind bekannt. Familiär gehäufte Fälle wurden beschrieben, wobei Mutationen der Z-Linie, Mitochondrien und des G4.5 Gens für Tafazzin isoliert werden konnten.
Glykogenspeichererkrankungen
Man unterscheidet PRKAG2 und Danon, eine Glykogenose Typ II.
Leitungsdefekte
Die Lenègre-Erkrankung ist ein primär progressiver Leitungsdefekt des His-Purkinje-Systems, der zur Verbreiterung des QRS-Komplexes im EKG führt mit langen Pausen, Bradykardie und Synkope.
Das Syndrom des kranken Sinusknotens (Sick-Sinus-Syndrom) gleicht phänotypisch einem Leitungsdefekt und kann autosomal-dominant auftreten.
Auch das Wolff-Parkinson-White-Syndrom (WPW) kommt selten familiär gehäuft vor.
Mitochondriale Myopathien
Teilweise werden die Enzyme der Atmungskette auf den Genen der Mitochondrien kodiert. Mehrere Syndrome aufgrund entsprechender Gendefekte sind bekannt, darunter das
- Kearns-Sayre-Syndrom mit Pigmentdegenerationen der Netzhaut des Auges, Augenmuskellähmung und Kardiomyopathie, und das
- MELAS-Syndrom mit Myopathie, Enzephalopathie, Laktatazidose und Schlaganfall-artigen Episoden. Außer den durch das Akronym definierten Merkmalen sind eine hypertrophe Kardiomyopathie und eine diffuse Koronargefäßerkankung typisch. Die Behandlung ist schwierig. Einige Patienten wurden mit kurzfristig bescheidenem Erfolg mit Coenzym Q behandelt. Eine Alternative stellt die Herztransplantation dar.
Ionenkanaldefekte
Es gibt eine wachsende Liste seltener erblicher und angeborener Herzrhythmusstörungen, die durch Gene für defekte Ionenkanalproteine kodiert werden. Auch ein kleiner Anteil von 5–10 % von Kindern mit dem plötzlichen Kindstod könnte durch Ionenkanaldefekte verursacht sein. Die klinische Diagnose eines Ionenkanaldefektes ist oft schon phänotypisch durch ein Standard-12-Kanal-EKG möglich. Einige dieser Fälle wurden zuvor als idiopathisches Kammerflimmern klassifiziert.
Long-QT-Syndrom (LQTS)
Das Long-QT-Syndrom ist wahrscheinlich die häufigste der Ionenkanalerkrankungen. Charakteristisch ist eine Verlängerung der Kammerdepolarisation und des QT-Intervalls im 12-Kanal-EKG. Es kommt zu einer speziellen Form der Kammertachykardie (torsade-de-pointes), und damit zu einem Risiko für Synkope und plötzlichen Herztod.
Jervell und Lange-Nielsen-Syndrom: selten, autosomal-rezessiv, assoziiert mit Taubheit. Zwei Gene, die einen langsam aktivierenden verzögerten Kaliumkanal kodieren.
Romano-Ward-Syndrom: viel häufiger, autosomal-dominant, acht verschiedene Gene, von denen sechs für verschiedene Kaliumkanäle kodieren, einer für einen Natriumkanal und einer für das Protein Ankryn, welches für die Verankerung von Ionenkanälen in der Zellmembran verantwortlich ist.
Brugada-Syndrom
Als klinische Entität ist das Brugada-Syndrom seit 1992 bekannt, es wird nach den beiden erstbeschreibenden Brüdern Pedro und Josep Brugada gelegentlich auch Brugada-Brugada-Syndrom genannt.[3] Es ist für den plötzlichen Herztod vor allem junger Menschen verantwortlich. Charakteristisch sind Rechtsschenkelblock-ähnliche Veränderungen im EKG, die ggf. durch einen Ajmalin-Test provoziert werden können. Verschiedene Gendefekte sind bekannt, sie wurden von Ramon Brugada, dem jüngsten der drei Brugada-Brüder beschrieben.
Asian SUNDS
SUNDS = „sudden unexplained nocturnal death syndrome“. Vornehmlich bei jungen asiatischen Männern, vor allem aus Thailand, Japan, den Philippinen und Kambodscha. Plötzlicher Tod im Schlaf durch Kammertachykardie oder Kammerflimmern. Einige Fälle sind vom Erscheinungsbild her nicht zu unterscheiden vom Brugada-Syndrom.
Short-QT-Syndrom (SQTS)
Erstbeschreibung im Jahre 2000. Das QT-Intervall im EKG ist verkürzt auf unter 330 ms. Es besteht eine große Gefahr für einen plötzlichen Herztod.
CPVT
CPVT = „Catecholaminergic Polymorphic Ventricular Tachycardia“, Erstbeschreibung durch Coumel 1978. Charakterisiert durch Synkope, plötzlichen Herztod, sowie durch große körperliche Anstrengung oder heftige Gefühlsbewegungen ausgelöste polymorphe Kammertachykardien bei Kindern und Jugendlichen. In 30 % plötzlicher Herztod bei Familienangehörigen. Verschiedene Vererbungsweisen sind bekannt.
Gemischte (angeborene und erworbene) primäre Kardiomyopathien
Dilatative Kardiomyopathie
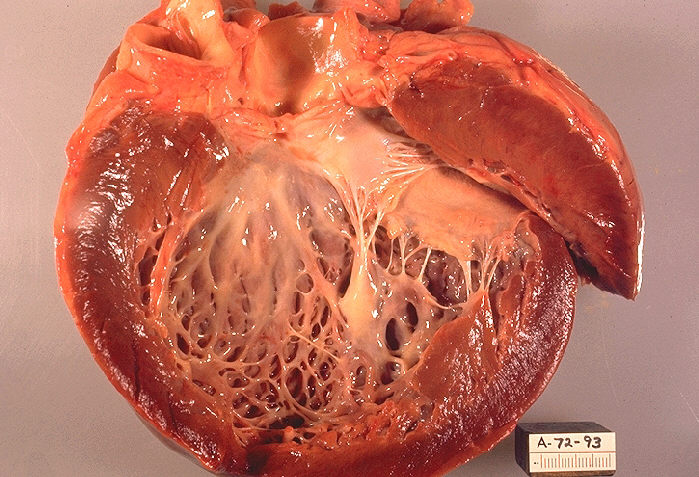
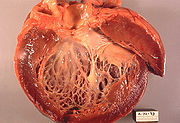 Dilatative Kardiomyopathie mit erweiterter linker Herzkammer. Eröffnetes Herz
Dilatative Kardiomyopathie mit erweiterter linker Herzkammer. Eröffnetes HerzDie dilatative Kardiomyopathie (DCM), bei der zunächst der linke Ventrikel (Herzkammer), im Endstadium auch alle Herzhöhlen, erheblich erweitert ist (das Herz kann mit einem großen, schlaffen Sack verglichen werden). Die Wandstärken sind meist nicht oder nur geringfügig verdickt (hypertrophiert). Das Herz zieht sich nur eingeschränkt zusammen (= systolische Funktionseinschränkung), oft kombiniert mit asynchronem Kontraktionsablauf der Kammer, bedingt durch eine Störung der Erregungsleitung infolge Linksschenkelblockes. Zahlenmäßig sind abgelaufene Herzmuskelentzündungen und chronischer Alkoholmissbrauch die häufigsten Ursachen. Es gibt auch angeborene Formen. Eine sekundäre Form stellt die „ischämische DCM“ in Folge einer koronaren Herzerkrankung dar, oder der Endzustand eines Hochdruckherzens. Die DCM ist ein häufiger Grund für eine Herztransplantation, wenn der Zustand des Patienten mit Medikamenten, Koronarintervention oder kardialer Resynchronisationstherapie (CRT) nicht ausreichend gebessert werden kann. Die Diagnose wird nach klinischem Verdacht mit den typischen Symptomen durch bildgebende Verfahren (Echokardiografie, MRT, MSCT) und feingeweblich (Myokardbiopsie) gesichert. Eine koronare Herzkrankheit muss durch eine Herzkatheteruntersuchung ausgeschlossen werden, da sich hieraus eine kurative Behandlungsmöglichkeit der Ursache ergeben könnte.
Restriktive Kardiomyopathie
Die restriktive Kardiomyopathie (RCM) stellt sich mit normal großen Herzkammern und einer meist normalen systolischen Pumpfunktion dar. Durch vermehrten Einbau von Bindegewebe in die Herzmuskulatur verhärtet die Herzmuskulatur. Die hierdurch versteiften Herzkammern lassen sich in der Erschlaffungsphase (Diastole) des Herzens schlecht füllen, das Blut staut sich in den Vorhöfen, die hierdurch stark vergrößert sind. Die Wanddicke der linken Herzkammer ist normal und die Herzklappen regelrecht.
Auffällig werden die Patienten durch Symptome einer Herzinsuffizienz wie lastabhängige Atemnot und Beinödeme. Die Erkrankung ist ausgesprochen selten, und kann übersehen werden, wenn man nicht gezielt danach fahndet. Diagnostische Methoden sind Echokardiografie, ggf. mit Gewebedoppler, Herzkatheteruntersuchung mit Hämodynamikmessung, ggf. Herzmuskelbiopsie, und MRT. Sporadische und familiär gehäufte Formen sind bekannt.
Erworbene primäre Kardiomyopathien
Myokarditis: entzündliche Kardiomyopathie
Die Herzmuskelentzündung ist ein akuter oder chronischer Prozess, der hervorgerufen werden kann durch eine große Bandbreite von
- Toxinen und Substanzen, z. B. Kokain, Interleukin-2
- infektiösen Erregern wie
- Viren, z. B. Coxsackie-Virus, Adenovirus, Parvovirus, HIV, die Beteiligung des Hepatitis-C-Virus ist umstritten [4]
- Bakterien, z. B. Diphtherie, Meningokokken, Psittakose, Streptokokken
- Rickettsien, z. B. Fleckfieber, Rocky-Mountain Spotted Fever
- Pilze, z. B. Aspergillus, Candida
- Parasiten, z. B. Trypanosoma cruzi (Chagas), Toxoplasmose
- der Whipple-Erkrankung (intestinale Lipodystrophie)
- Riesenzellmyokarditis
- Überempfindlichkeitsreaktionen auf Medikamente wie Antibiotika, Sulfonamide, Antikonvulsiva und Antirheumatika.
Die endokardiale Fibroelastose bei Neugeborenen und Kleinkindern ist das Resultat einer intrauterinen Infektion mit dem Mumpsvirus.
Stressprovoziert (Tako-Tsubo)
Tako-Tsubo-Kardiomyopathie, Stress-Kardiomyopathie, Broken-Heart-Syndrom oder apical ballooning wird eine Herzmuskelerkrankung genannt, die meist bei postmenopausalen Frauen und häufig nach emotionalen Stresssituationen auftritt, und sowohl in Beschwerdebild, EKG-Veränderungen als auch Laborwerten wie ein akuter Herzinfarkt imponiert. Es zeigt sich eine ballonartige Auftreibung der Herzkammerspitze wie bei einem schweren Vorderwandinfarkt, aber es finden sich keinerlei Engstellen oder Verschlüsse der Herzkranzgefäße. Als Ursache wird eine Stresshormon-bedingte, nur vorübergehende Verschließung der feinen Haargefäße der Herzkranzgefäße angenommen. Hierdurch kommt es zu einer vorübergehenden „Schockstarre“ (stunning) des Herzmuskels, der anders als beim echten Herzinfarkt nicht abstirbt (Nekrose), sondern sich wieder völlig erholen kann. Die Prognose ist in der Regel gut und in einigen Monaten ist die Herzmuskelstörung rückläufig. Die Sterblichkeit beträgt etwa 3 %. Die Diagnostik erfolgt durch Echokardiografie, Herzkatheteruntersuchung und Magnetresonanztomografie (MRT).
Schwangerschafts-Kardiomyopathie
Seltene dilative K. mit systolischem Herzversagen bei Schwangeren im letzten Trimenon oder bis 5 Monate nach Entbindung (peripartale K.). Die Ursache ist nicht bekannt. Meist sind übergewichtige Schwangere über 30 Jahren betroffen, die bereits mehrfach entbunden haben und eine Präeklampsie hatten. Etwa die Hälfte der Patientinnen haben sich nach einem halben Jahr nahezu erholt, in Einzelfällen kommt es jedoch zu fortschreitendem Herzversagen mit Tod oder Herztransplantation.
Tachykardie-induziert
auch Tachymyopathie genannt. Meist reversibel, tritt im Anschluss an eine länger anhaltende tachykarde Herzrhythmusstörung auf.
Neugeborene insulin-abhängiger diabetischer Mütter
Sekundäre Kardiomyopathien
Die wichtigsten und häufigsten der überaus zahlreichen sekundären K. sind hier aufgelistet, wobei es sich teils um angeborene, teils um erworbene Erkrankungen handelt.
Infiltrativ
Hier werden die Amyloidose, der Morbus Gaucher, Morbus Hurler und Morbus Hunter unterschieden.
Speichererkrankungen
Die Hämochromatose, Morbus Fabry, Glykogenspeicherkrankheit vom Typ II (Morbus Pompe) und der Morbus Niemann-Pick sind zu erwähnen.
Toxisch
Als Kardiotoxizität beschreibt die schädigende Wirkung einer Substanz (Medikamente, Drogen, Schwermetalle und Chemikalien) oder eines Krankheitserregers auf den Herzmuskel. Hieraus kann Kardiomyopathie resultieren.
Endomyokardial
Hier ist die Endomyokardfibrose zu nennen, bei der sich das Endokard (Herzinnenhaut) porzellangussartig verdickt hat und zu einer diastolischen Herzfunktionsstörung führt, sowie das hypereosinophile Syndrom (Löffler-Syndrom).
Entzündlich – granulomatös
Endokrin
Bei Diabetes mellitus, Funktionsstörungen der Schilddrüse (Hyperthyreose, Hypothyreose) und Nebenschilddrüse (Hyperparathyroidismus), beim Phäochromozytom und der Akromegalie sind Beteiligungen der Herzmuskulatur im Sinne einer K. beschrieben.
Kardiofazial
Noonan-Syndrom und Lentiginose
Neuromuskulär / neurologisch
Friedreich-Ataxie, Muskeldystrophien nach Duchenne-Becker und Emery-Dreifuss, Myotone Dystrophie, Neurofibromatose und Tuberöse Sklerose können mit einer K. einhergehen.
Mangelernährung
Beriberi (Thiamin), Pellagra (Vitamin B), Skorbut (Ascorbinsäure), Selenmangel (Keshan-Krankheit), Karnitinmangel und Kwashiorkor können eine K. verursachen.
Autoimmun / Bindegewebe
Oft übersehen wird die Herzbeteiligung bei Erkrankungen aus dem rheumatischen Formenkreis: Systemischer Lupus erythematodes, Dermatomyositis, Rheumatoide Arthritis, Sklerodermie und Polyarteriitis nodosa.
Elektrolytstörungen
Folgen einer Krebstherapie
Anthrazycline: Doxorubicin (Adriamycin), Daunorubicin, Cyclophosphamid und Bestrahlungstherapie können den Herzmuskel schädigen.
Kardiomyopathie bei Haustieren
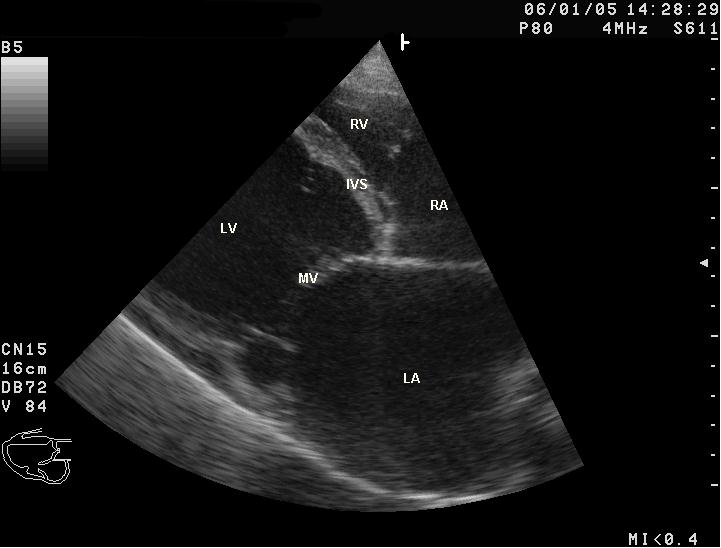
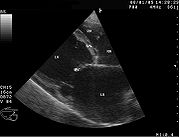 Sonographische Darstellung der DCM beim Hund mit stark vergrößertem Herzen. LV-linke Herzhauptkammer; RV-rechte Herzhauptkammer; LA-linker Vorhof, RA-rechter Vorhof, IVS-Herzscheidewand, MV-Mitralklappe
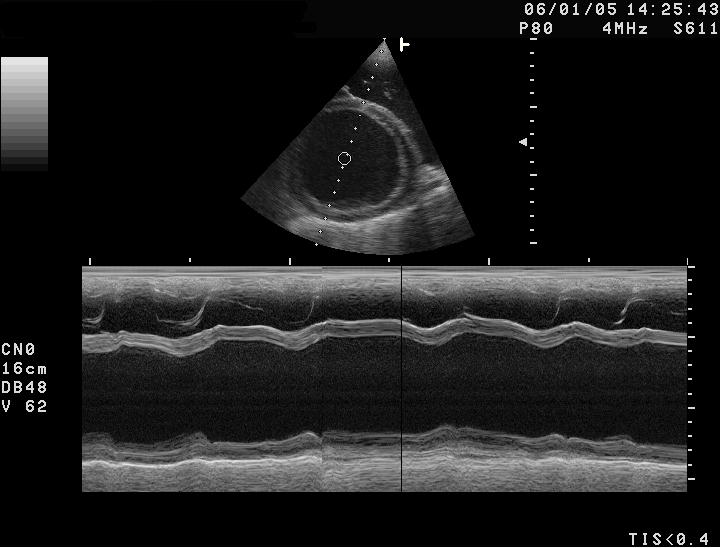
Sonographische Darstellung der DCM beim Hund mit stark vergrößertem Herzen. LV-linke Herzhauptkammer; RV-rechte Herzhauptkammer; LA-linker Vorhof, RA-rechter Vorhof, IVS-Herzscheidewand, MV-Mitralklappe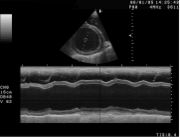 M-Mode-Darstellung der DCM. Der Herzmuskel zeigt eine Kontraktilität von etwa 10 % (normal: >25 %) und ist stark erweitert.
M-Mode-Darstellung der DCM. Der Herzmuskel zeigt eine Kontraktilität von etwa 10 % (normal: >25 %) und ist stark erweitert.Bei Kleintieren zählen Kardiomyopathien zu den am häufigsten beobachteten Herzerkrankungen. Die Ausprägungen unterscheiden sich je nach Tierart und Rasse erheblich.
Hunde
Bei Hunden ist die dilatative Kardiomyopathie (kurz DKMP oder DCM) die dominierende Form dieser Herzerkrankung. Vor allem die Vertreter größerer Rassen neigen zu diesem Leiden, während Angehörige kleinerer Hunderassen häufiger von Degenerationserscheinungen des Endokards im Klappenbereich betroffen sind. Einen einfachen Anhaltspunkt gibt hier die sogenannte „Ein-Hand-Regel“. Kann der Hund mit einer Hand angehoben werden, hat er wahrscheinlich keine Kardiomyopathie.
Die Erkrankung verläuft in unterschiedlichen Formen. Wird bei einem Dobermann-Pinscher die Erkrankung diagnostiziert, beträgt seine wahrscheinliche restliche Lebenszeit weniger als ein halbes Jahr. Andere Rassen wie Neufundländer oder Deerhounds zeigen einen wesentlich milderen Verlauf. Eine Sonderform stellt die dilatative Kardiomyopathie des Boxers dar, die eher an die arrhythmogene rechtsventrikuläre Kardiomyopathie des Menschen erinnert.
Behandlung: Da eine grundlegende Therapie wie beim Menschen mittels Herztransplantation aus ethischen und finanziellen Gründen kaum durchgeführt wird, beschränkt sich die Behandlung auf die Verabreichung kontraktionsfördernder Medikamente (Pimobendan, Digoxin) sowie die medikamentelle Milderung der Folgeerscheinungen (Herzrhythmusstörungen, Lungenödem etc.). Da für einige Rassen die Erblichkeit der Erkrankung nachgewiesen wurde, haben einige Zuchtvereine Zuchtuntersuchungen initiiert.
Katzen
Bei Katzen ist die hypertrophe Kardiomyopathie die am häufigsten beobachtete Form der Erkrankung. Sie wird auch als Folgeerscheinung einer Schilddrüsenüberfunktion beobachtet (thyreotoxische Kardiomyopathie). Eine Sonderform gleicht der hypertroph-obstruktiven Kardiomyopathie des Menschen. Daneben sind auch die restriktive und die dilatative Kardiomyopathie bei Katzen beschrieben. Letztere wird aber als sekundäre Kardiomyopathie eingestuft, da sie die Folgeerscheinung eines Mangels an dem für Katzen essentiellen Taurin ist. Da industrielle Fertigfuttermittel mit diesem Inhaltsstoff angereichert sind, wird diese Form der Erkrankung bei Katzen zunehmend selten beobachtet. Weiterhin existieren Übergangsformen, die sowohl Zeichen einer Erweiterung als auch einer Hypertrophie zeigen. Sie werden als intermediäre Kardiomyopathie bezeichnet.
Ähnlich wie bei Hunden beschränkt sich die Therapie der Erkrankung bei Katzen auf eine Medikamentengabe zur Verbesserung der Symptomatik (Verbesserung der systolischen oder diastolischen Funktion, Behandlung von Rhythmusstörungen) beziehungsweise auf die Therapie der Grunderkrankung (v. a. Schilddrüsenerkrankungen und Taurinsubstitution).
Zur Behandlung der Kardiomyopathie bei Katzen werden Entwässerungsmedikamente (Diuretika), Beta-Blocker und ACE-Hemmer eingesetzt. Sie können den Krankheitsverlauf positiv beeinflussen und je nach Stadium der Krankheit dem Tier noch viele Monate bis Lebensjahre ermöglichen.
Literatur
- Crawford MH, DiMarco JP, Paulus WJ (eds.): Cardiology 2nd ed., Mosby, Edinburgh 2004 ISBN 0-323-02405-X
- Dietel M, Suttorp N, Zeitz M (Hrsg); Kasper DC, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL (eds.): Harrisons Innere Medizin. Deutsche Ausgabe in Zusammenarbeit mit der Charité. McGraw Hill, ABW-Wissenschaftsverlag, 16. A., Berlin 2005 ISBN 3-936072-29-9
- Fuster V, Alexander W, O’Rourke RA (eds.): Hurst’s The Heart. 11th ed., McGraw-Hill, New York 2004 ISBN 0-07-142264-1
- Topol EJ (ed.): Textbook of Cardiovascular Medicine. 2nd ed., Lippincott Williams & Wilkins, Philadelphia 2002 ISBN 0-7817-3225-5
- Werdan K, Trappe HJ, Zerkowski HR (Hrsg.): Das Herz-Buch. Praktische Herz-Kreislauf-Medizin. Urban & Fischer, München Jena 2003 ISBN 3-437-21790-9
- Zipes DP, Libby P, Bonow RO, Braunwald E: Braunwald’s Heart Disease, A Textbook of Cardiovascular Medicine. Elsevier Saunders, Philadelphia 2005. ISBN 0-8089-2305-6
Quellen
- ↑ Maron BJ et al.: Contemporary Definitions and Classification of the Cardiomyopathies. Ein "Scientific Statement" der American Heart Association. Circulation (2006) 113:1807-1816. PMID 16567565 Originalartikel als PDF-Datei
- ↑ Thiene G, Corrado D, Basso C: Arrhythmogenic right ventricular cardiomyopathy/dysplasia. Orphanet J Rare Dis. 2007 Nov 14;2:45. PMID 18001465
- ↑ Brugada P, Brugada J: Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report. J Am Coll Cardiol (1992) 20(6):1391-6. PMID 1309182.
- ↑ Japanische Studie mit positivem Ergebnis für Hepatitis C, Multizentrische Studie ohne Hepatitis-C-Nachweis

Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.