- Marfan-Syndrom
-
Klassifikation nach ICD-10 Q87.4 Marfan-Syndrom ICD-10 online (WHO-Version 2011) Das Marfan-Syndrom ist eine systemische Besonderheit des Bindegewebes auf der Grundlage einer Genmutation; sie kann autosomal-dominant vererbt werden oder vereinzelt (als Neumutation) auftreten.
Inhaltsverzeichnis
Auftrittshäufigkeit
Die Krankheit tritt mit einer Häufigkeit von etwa 1:5.000 bis 1:10.000 auf, wobei sechs bis sieben von zehn Fällen familiär bedingt sind. Der Anteil der Neumutationen beträgt 25 bis 40 %. Da die Definition „Marfan-Syndrom“ derzeit nach Hilfskriterien erfolgt und es mehrere verwechselbare „Parallelkrankheiten“ gibt, sollte besser vom Marfan-Phänotyp gesprochen werden. Dafür insgesamt ist die Häufigkeit etwa 1:6.000. Die Gruppe der Krankheiten des Marfan-Phänotyps wird neuerdings „Fibrillopathien“ genannt.
Erstbeschreibung
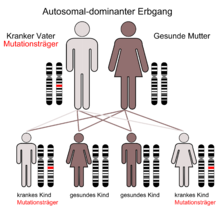 Der autosomal-dominante Erbgang
Der autosomal-dominante Erbgang
Das Syndrom wurde erstmals unter wissenschaftlichen Gesichtspunkten 1896 von dem französischen Kinderarzt Antoine Marfan (1858-1942) beschrieben. In den 1930er Jahren erkannte man, dass die Besonderheit nicht geschlechtsgebunden (gonosomal) vererbt wird, sondern dass Veränderungen auf dem Chromosom 15 ursächlich sind (autosomal-dominanter Erbgang).
Krankheitsursache
Das Gen für das Marfan-Syndrom liegt auf dem langen Arm des Chromosom 15 (15q21) und ist inzwischen sequenziert. Eine Mutation des Fibrillin-Genes (Fibrillin-1-Gen=FBN1) führt zu einem verkürztem Genprodukt oder einer Missense-Mutation, Mutation des TGF-β-II-Rezeptors.
Merkmale
Feingeweblich manifestiert sich das Marfan-Syndrom in der Feinstruktur des Bindegewebes (der Mikrofibrillen). Das Bindegewebe ist also fehlerhaft aufgebaut und führt zu einer mehr oder weniger ausgeprägten Instabilität aller Bindegewebe des Körpers.
Folgende Symptome kommen häufig bei Menschen mit Marfan-Syndrom vor, wobei nicht alle Merkmale bei allen Menschen oder bei allen Menschen in gleich starker Ausprägung vorliegen:
1. Herz- und Gefäßsystem
- Herzklappenfehler (Schlussunfähigkeit der Aorten- oder Mitralklappe - Aneurysma der Aortenwurzel)
- Aortendissektion (Aufspaltung oder Zerreißung der Aortenwand)
2. Augen
- Lockerung des Halteapparates der Linsen (Linsenluxation)
- Kurzsichtigkeit
- Glaukom (Grüner Star = hoher Augeninnendruck)
- Katarakt (Grauer Star = Linsentrübung)
- Netzhautablösung: Hierdurch kommt es zu dem Symptom der Lichtblitze (Differentialdiagnose)
- blaue Skleren
- eventuell Iridodonesis (Irisschlottern bei Kopfbewegung)
- senkrechte Überentwicklung des Kopfes (Dolichocephalie / Langschädel)
- Hyperlaxizität (Überstreckbarkeit) der Gelenke
- hoher Gaumen (Gotischer Gaumen)
- Arachnodaktylie (Spinnenfingrigkeit); verschmälerte Finger (Madonnenfinger)
- Trichter- oder Kielbrust
- Hochwuchs
- Verformungen der Wirbelsäule (Skoliose, Hyperkyphose)
- schwach entwickelte Muskulatur (Muskelhypotrophie)
- Plattfüße
- samtartige Haut mit Neigung zu Striae (Dehnungsstreifen)
- Neigung zum Leistenbruch
- Neigung zu atlantoaxialer oder zervikookzipitaler Instabilität, dadurch unter anderem erhöhte Anfälligkeit für das Grisel-Syndrom
4. Innere Organe
- die Bindegewebsschwäche kann auch die Lunge angreifen, wodurch ein so genannter Pneumothorax auftreten kann
Diagnose und Behandlung
Die Diagnostik und Behandlung sollte stets fachübergreifend (unter anderem durch Kinderkardiologen / Kardiologen, Orthopäden, Augenärzte) erfolgen gemäß der Genter Nosologie (systematische Beschreibung der Krankheiten).
Bislang existiert noch keine ursächlich heilende Therapie.
Einen für die Zukunft denkbaren ursächlichen Therapieansatz zeigen die Ergebnisse einer im April 2006 im Wissenschaftsmagazin Science publizierten Studie, die in einem Mausmodell des Marfan-Syndroms das Zytokin TGF-β mit der typischen Entwicklung von Aortenaneurysmen und -dissektionen in Verbindung bringen konnte. Durch eine Behandlung mit dem als Blutdrucksenker bereits in der Humanmedizin verwandten AT1-Antagonisten Losartan konnten die Mäuse wirksam vor der Ausprägung dieser lebensgefährlichen Veränderungen der Gefäßwand geschützt werden, da Losartan die Wirkung des überaktiven TGF-β antagonisiert. Bereits bestehende Veränderungen am Herzen normalisierten sich zudem weitestgehend, solche in anderen Bereichen des Körpers zumindest teilweise.[1]
Lebenserwartung
Die größte Bedeutung zur deutlichen Verlängerung der Lebenserwartung fällt in den Bereich der Kinderkardiologie / Kardiologie. Es gibt Hinweise darauf, dass die Geschwindigkeit der Entstehung einer Aortenerweiterung durch die Einnahme von Beta-Blockern vermindert werden kann. Wie weit eine frühe und prophylaktische Einnahme im Kindesalter eine Aortenerweiterung verhindert, ist noch unklar; erste Erfahrungen nach 14 Jahren Beta-Blocker-Therapie bei Kindern (2005), begonnen bei drei- bis vierjährigen betroffenen Kindern zeigen aber, dass eine Progredienz der Erweiterung der Aortenwurzel in fast allen Fällen nicht eintritt und die Erweiterung unter der Medikation dann nur altersspezifisch erfolgt. Ist die Aortenwurzel bei Erwachsenen auf mehr als 55 mm erweitert, muss eine Operation erfolgen, um eine Aortendissektion oder -ruptur zu verhindern. Dies geschieht evtl. durch einen Aortenwurzelersatz mit künstlicher Herzklappe. Die postoperative Überlebensrate nach 10 Jahren beträgt zwischen 70 und 75 %. Bei Kindern leitet sich die Notwendigkeit einer Operation aus der Abweichung des aktuellen Aortendurchmessers vom altersbezogenen Normalwert ab, der individuell geprüft werden muss. Entscheidend ist, dass bei Kindern und Erwachsenen die Endokarditis-Prophylaxe ganz besonders ernst genommen wird (selbst bei zahnärztlichen Eingriffen, kleinen Wunden, wund gelaufenen Füßen usw.), da eine Besiedlung/Vegetation an den besonders sensiblen Herzklappen und Klappenrändern ein Voranschreiten der marfan-typischen Gewebeinstabilität auslösen kann.
Bei schweren Verläufen können auch augenärztliche Eingriffe zum Erhalt oder Wiederherstellung der Sehfähigkeit erforderlich werden. Bei Überdehnung und zunehmendem Verlust (Einreißen) der Zonulafasern entsteht das so genannte „Linsenschlottern“. Schlotterlinsen müssen nicht operiert werden, nur das Verschieben der Linsen kann den Augendruck so stark werden lassen, dass die Entfernung der Linse notwendig wird. Es sind inzwischen neue OP-Techniken entwickelt worden, die unter Erhalt des Kapselsackes und Implantation einer Speziallinse bereits bei kleinen Kindern die Sehfähigkeit erhalten kann.
Bei Verläufen mit schweren Wirbelsäulenverbiegungen sind Aufrichtungsoperationen wahrscheinlich. Es sind neue OP-Techniken entwickelt worden, die eine Aufrichtung der Wirbelsäule ohne Vollversteifung ermöglichen.
In den siebziger Jahren ermittelte eine Studie die Lebenserwartung eines Menschen mit Marfan-Syndrom mit 30 bis 40 Jahren. Seit dieser Zeit wurden sowohl die Diagnostik als auch die medikamentöse und chirurgische Behandlung optimiert, so dass die Lebenserwartung sich annähernd der Normalbevölkerung angeglichen hat. Trotzdem sterben noch immer viele der Betroffenen, weil die rettende Diagnose nicht gestellt wurde.
Literatur
- Arslan-Kirchner, Mine; Kodolitsch, Yskert von; Schmidtke, Jörg: Genetische Diagnostik beim Marfan-Syndrom und verwandten Erkrankungen: Bedeutung des klinischen Managements, Dtsch Arztebl 4. Juli 2008; 105(27): 483, DOI: 10.3238/arztebl.2008.0483, Übersichtsarbeit
- Marfanhilfe (Deutschland) e. V. Herausgeber: Marfan-Syndrom, Ein Ratgeber für Patienten, Angehörige und Betreuende, Steinkopf, Darmstadt ISBN 3-7985-1565-4
- Marfan Stiftung Schweiz (Hrsg.): Herzsache. Gesundheitskompetenz und Empowerment am Beispiel des Marfan-Syndroms, Bern 2008, ISBN 978-3-033-01587-6
- Witkowski, Prokop, Ullrich, Thiel: Lexikon der Syndrome und Fehlbildungen (7. Auflage, 2003)
Weblinks
- Marfan Hilfe (Deutschland) e.V. Verein für Menschen mit Marfan Syndrom oder anderen mikrofibrillären Erkrankungen
- Marfan Stiftung Schweiz Information, Dokumentation und Beratung für Menschen mit Marfan-Syndrom und verwandten Bindegewebeschwächen
- Marfan Syndrome (engl.) Webseite des Cold Spring Harbor Laboratory mit vielen Animationen und Videos
Quellen
- ↑ Habashi, J. et al. (2006): Losartan, an AT1 Antagonist, Prevents Aortic Aneurysm in a Mouse Model of Marfan Syndrome. Science, 312: 117 - 121 (Abstract) (Material und Methoden + sehr beeindruckende Bilder PDF; 462 KB)

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Genetische Störung
- Krankheitsbild in der Kinderkardiologie
- Krankheitsbild in der Inneren Medizin
Wikimedia Foundation.