- Proteinkristallographie
-
Kristallstrukturanalyse ist die Bestimmung des atomaren Aufbaus eines Kristalls durch Beugung geeigneter Strahlung am Kristallgitter. Sehr häufig wird hierfür monochromatische Röntgenstrahlung verwendet, da sich diese verhältnismäßig einfach als charakteristische Röntgenstrahlung einer Röntgenröhre erzeugen lässt. Hierfür hat sich der Begriff Röntgenstrukturanalyse eingebürgert. Alternativ lassen sich auch Neutronenstrahlen oder Synchrotronstrahlung verwenden. Die Kristallstrukturanalyse mit Elektronenstrahlen ist aufgrund der starken Wechselwirkung zwischen den eingestrahlten Elektronen und dem Kristall besonders schwierig und für Routineuntersuchungen noch nicht ausgereift.
Aus dem beobachteten Beugungsmuster kann anschließend die Kristallstruktur berechnet werden. Die Geometrie der Elementarzelle des Kristallgitters kann vollständig anhand der Winkel abgeleitet werden, unter denen die Beugungsmaxima auftreten. Aus der Stärke der Beugungsmaxima kann mittels verschiedener mathematischer Methoden die Anordnung der Atome innerhalb der Elementarzelle berechnet werden. Die hierbei benötigten Rechnungen werden allerdings bereits für mittelgroße Moleküle (ab etwa 10 Nicht-Wasserstoffatomen) so komplex, dass sie ohne Computer nicht durchführbar sind. Daher können Proteinstrukturen erst seit den 60er Jahren, nach der Entwicklung des Computers, analysiert werden, obwohl Forscher bereits 1934 herausfanden, dass das Enzym Pepsin regelmäßige Kristalle bildet.
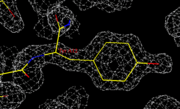 Bei der Proteinkristallographie wird die Struktur ermittelt, indem die Aminosäuresequenz in die Elektronenverteilung (weißes Gitter) eingepasst und modifiziert oder verschoben wird, bis plausibel ist, dass die gewählte Struktur die ermittelte Elektronenverteilung erzeugen kann.
Bei der Proteinkristallographie wird die Struktur ermittelt, indem die Aminosäuresequenz in die Elektronenverteilung (weißes Gitter) eingepasst und modifiziert oder verschoben wird, bis plausibel ist, dass die gewählte Struktur die ermittelte Elektronenverteilung erzeugen kann.Bei der Kristallstrukturanalyse mittels Röntgen-, Elektronen- oder Synchrotronstrahlung werden streng genommen nicht die Positionen der Atome, sondern wird die Verteilung der Elektronen in der Elementarzelle bestimmt, da diese mit der Strahlung in Wechselwirkung treten. Man erhält also eigentlich eine Elektronendichtekarte, und bei sehr exakten Kristallstrukturanalysen von Molekülen mit leichten Atomen findet man in der Tat Bindungselektronen. Neutronen treten dagegen mit dem Atomkern in Wechselwirkung. Allerdings ist der Unterschied in der Position in den meisten Fällen vernachlässigbar. Eine genaue Beschreibung der Beugungseffekte an Kristallen und deren Interpretation ist im Artikel Röntgenbeugung zu finden.
Idealerweise wird die Beugung an einem Einkristall durchgeführt. Häufig ist dies aber nicht möglich, da nicht immer genügend große Einkristalle einer Substanz zur Verfügung stehen. Heutzutage ist es möglich, auch das Beugungsmuster von Kristallpulvern im Rahmen einer Kristallstrukturanalyse auszuwerten (Rietveld-Methode). Allerdings geht durch die hierbei auftretende Überlagerung von Beugungsmaxima Information verloren, so dass die Ergebnisse im allgemeinen von geringerer Qualität sind.
Neben der eigentlichen kristallographischen Anwendung der Methode, bei welcher der Kristall selber von Interesse ist, wird die Kristallstrukturanalyse auch zur Aufklärung von Molekülstrukturen verwendet. Dies ist heute eine Standardmethode der Chemie und der Biochemie und damit ein Teilgebiet der Strukturbiologie. Hierfür ist allerdings die Kristallisation der Moleküle Voraussetzung, was insbesondere bei Proteinkristallen sehr schwierig sein kann. Durch Kristallisation in Gegenwart von Substraten kann versucht werden, verschiedene metabolische Zustände des Proteins zu erfassen. Ein weiteres Problem hierbei besteht darin, dass die Proteinstruktur sich durch die Kristallisation möglicherweise ändert. Da man von der Struktur auf die Funktion des Proteins sowie auf mögliche Liganden (z. B. Medikamente) schließen möchte, können solche Strukturänderungen problematisch für die Anwendung in Medizin und Forschung sein.
Eine konkurrierende Methode zur Strukturbestimmung von Proteinen mittels Röntgenkristallstrukturanalyse stellt die NMR-Spektroskopie dar, die allerdings derzeit nur für Proteine kleiner oder mittlerer Größe verwendet werden kann.
Auf dem Gebiet der Kristallstrukturanalyse gab es mehrere Nobelpreise, angefangen bei Max von Laue und Wilhelm Conrad Röntgen, die die Grundlagen legten, über zum Beispiel Dorothy Crowfoot Hodgkin, die viele biologisch relevante Moleküle erstmalig strukturell bestimmte, bis zu Robert Huber und Johann Deisenhofer, die Proteine (unter anderem auch das chlorophyllhaltige Photoreaktionszentrum ) als Proteinkristalle untersuchten. Eines der bekanntesten Beispiele für die Strukturaufklärung mittels Röntgenbeugung ist die Entschlüsselung der DNA-Struktur durch James Watson und Francis Crick, deren Modell wesentlich auf Röntgenbeugungsdaten von Maurice Wilkins und Rosalind Franklin beruhte. 1985 wurde Jerome Karle und Herbert A. Hauptman der Nobelpreis für Chemie für deren Beiträge zur Entwicklung der "Direkten Methoden" zur Kristallstrukturanalyse zuerkannt.
Literatur
- Massa, Werner: Kristallstrukturbestimmung, Stuttgart: Teubner 2002, ISBN 3519135272
- C. Giacovazzo: Fundamentals of Crystallography, Oxford University Press; ISBN 0-19-855579-2
Weblinks

Wikimedia Foundation.