- Chronisch inflammatorische demyelinisierende Polyneuropathie
-
Die chronisch inflammatorische demyelinisierende Polyradikuloneuropathie (CIDP) ist eine sehr selten auftretende entzündliche Erkrankung der peripheren Nerven (Polyradikulitis), die sich durch eine allmählich zunehmende Schwäche in den Beinen und mitunter auch Armen bemerkbar macht. Diese ansteigenden Schwächezustände entwickeln sich über einen Zeitraum von zwei Monaten oder länger, was das hauptsächliche diagnostische Kriterium zur Abgrenzung gegen das Guillain-Barré-Syndrom darstellt.
Die Erkrankung beruht auf einer Schädigung der Myelinschicht, die die Nervenfortsätze umkleidet. Die chronisch inflammatorische demyelinisierende Polyneuropathie ist behandelbar. Seit der Erstbeschreibung 1890 wurden anhand mehrerer Studien die klinischen Manifestationen und Diagnosekriterien bestimmt, die untereinander nur gering variieren.
Inhaltsverzeichnis
Epidemiologie
Die chronisch inflammatorische demyelinisierende Polyneuropathie kann sowohl männliche als auch weibliche Personen jeden Alters befallen. Männer sind etwa doppelt so häufig betroffen wie Frauen. Möglicherweise wird sie jedoch zu selten diagnostiziert, denn die Krankheitshäufigkeit (Prävalenz) liegt bei 1-2/100.000, wobei ein Erkrankungsgipfel im 5. - 6. Lebensjahrzehnt liegt.
Entstehung (Pathogenese) von CIDP
Gegenwärtige Theorien vermuten, dass das Immunsystem des Körpers, das ihn normalerweise vor Krankheitserregern schützt, Komponenten der Myelinschicht als Fremdstoff empfindet und abwehrt. Unklar ist jedoch, was diesen Vorgang auslöst.
Bei einigen Patienten enthält das Blut abnormale Eiweißstoffe, welche die Schädigung fördern. Derzeitige pathogenetische Konzepte legen eine abweichende (aberrierende) Immunantwort auf zellulärer und humoraler Ebene nahe, die sich gegen Antigene der peripheren Nerven richtet und bei der Antikörper, Komplement, Cytokine, autoreaktive T-Zellen und Makrophagen eine Rolle spielen. Im Gegensatz zum Guillain-Barré-Syndrom (GBS) geht einer CIDP nur selten eine Infektion voraus.
Die CIDP tritt häufig in Assoziation mit anderen Erkrankungen auf, zum Beispiel HIV-Infektionen, systemischem Lupus erythematodes, Diabetes mellitus, Hepatitis C, Paraproteinämien und malignen Erkrankungen wie Lymphom und osteosklerotischem Myelom.
Klinische Manifestation
Die CIDP entwickelt sich langsam und erreicht ihr Maximum definitionsgemäß acht Wochen und später nach Symptom-Beginn. Es kommt zu symmetrischen Lähmungen (Paresen), proximal (zur Körpermitte hin) oder distal (von der Körpermitte weg) mit Reflexabschwächung oder -verlust und unterschiedlicher sensibler Beteiligung. Häufig klagen Patienten über Müdigkeit, Sensibilitätsstörungen (Parästhesien), die sich durch ein Kribbeln oder Brennen bemerkbar machen, oder Kompressionsgefühlen an den Extremitäten. Lähmungen an der oberen Extremität verursachen eine eingeschränkte Feinmotorik.
Lähmungen an der unteren Extremität führen zu einem Verlust der Knie- und Fußgelenkreflexe, Gehstörungen, Schwierigkeiten beim Treppensteigen und Aufstehen von Sitzgelegenheiten. Eine sensible Gangataxie (breitbeiniger, schwankender, unsicherer Gang) kann vorhanden, aber auch, vor allem bei Kindern, alleiniges Symptom sein. Beschwerden beim Wasserlassen können ebenfalls auftreten. Aus noch unklaren Gründen kommt es gelegentlich zum Tremor (Zittern).
Die Häufigkeitsverteilung der neurologischen Defizite:
- Motorische Defizite mit 94 %
- Parästhesien mit 64 %
- Hirnnerven-Beteiligung in 2-32 %
Varianten der CIDP
- Die sensorische CIDP: charakterisiert durch überwiegend sensible Symptome oder eine unregelmäßige (ataktische) Neuropathie. Elektrophysiologisch zeigt sich eine Beteiligung der motorischen Nerven, nach längerem Verlauf sind auch motorische Ausfälle nachzuweisen.
- Lewis-Sumner-Syndrom: ist eine multifokale erworbene demyelinisierende sensorische und motorische Neuropathie (MADSAM). Diese zeigt eine asymmetrische Verteilung, tritt anfänglich an der oberen Extremität auf (vorwiegend sensorisch oder sensomotorisch). Charakteristisch ist der Nachweis multifokaler Leitungsblöcke in der Elektrophysiologie und ein gutes Ansprechen auf intravenöse Immunglobuline. In der Literatur finden sich Übereinstimmungen mit Patienten mit fokaler demyelinisierender Neuropathie an der oberen Extremität.
- CIDP mit zus. monoklonaler IgM-Gammopathie und Nachweis von Antikörpern gegen Myelin-Glykoprotein (MAG-AK)
- Die CIDP mit MGUS (monoklonale Gammopathie unbestimmter Signifikanz): ca. 10-20 % der Patienten mit typischer CIDP haben eine monoklonale IgG- oder IgA-Gammopathie unbestimmter Signifikanz. Die klinische Manifestation und die Therapie-Reaktionen sind ähnlich wie bei CIDP-Patienten ohne Paraproteinämie.
- Axonale Varianten: Fälle von chronisch-relapsierender und progredienter axonaler Polyneuropathie (steroidresponsibel) sind bekannt. Bis heute fehlt jedoch der Beweis, dass es sich um immunvermittelte Neuropathien handelt. Auch ist unklar, ob es eine Assoziation gibt zu einer erworbenen multifokalen sensomotorischen Neuropathie, charakterisiert durch eine asymmetrische axonale Neuropathie mit Nachweis von Gangliosid-Antikörpern.
Diagnostik
Die Elektroneurographie erbringt den Nachweis von verlangsamten Nervenleitgeschwindigkeiten (Demyelinisierung) auf weniger als 70-80 % des unteren Normwertes, verlängerten distalen Latenzen, verlängerter F-Wellen-Latenz oder F-Wellen-Verlust. In der Liquordiagnostik zeigt sich eine Eiweißerhöhung (Schrankenstörung), die allerdings völlig unspezifisch ist. Mittels Magnetresonanztomographie lassen sich unter Umständen symmetrisch verteilte entzündliche Veränderungen und/oder Verdickung spinaler Wurzeln nachweisen. Labordiagnostisch ist der Nachweis von Gangliosid-Antikörpern im Serum möglich.
Nervenbiopsie
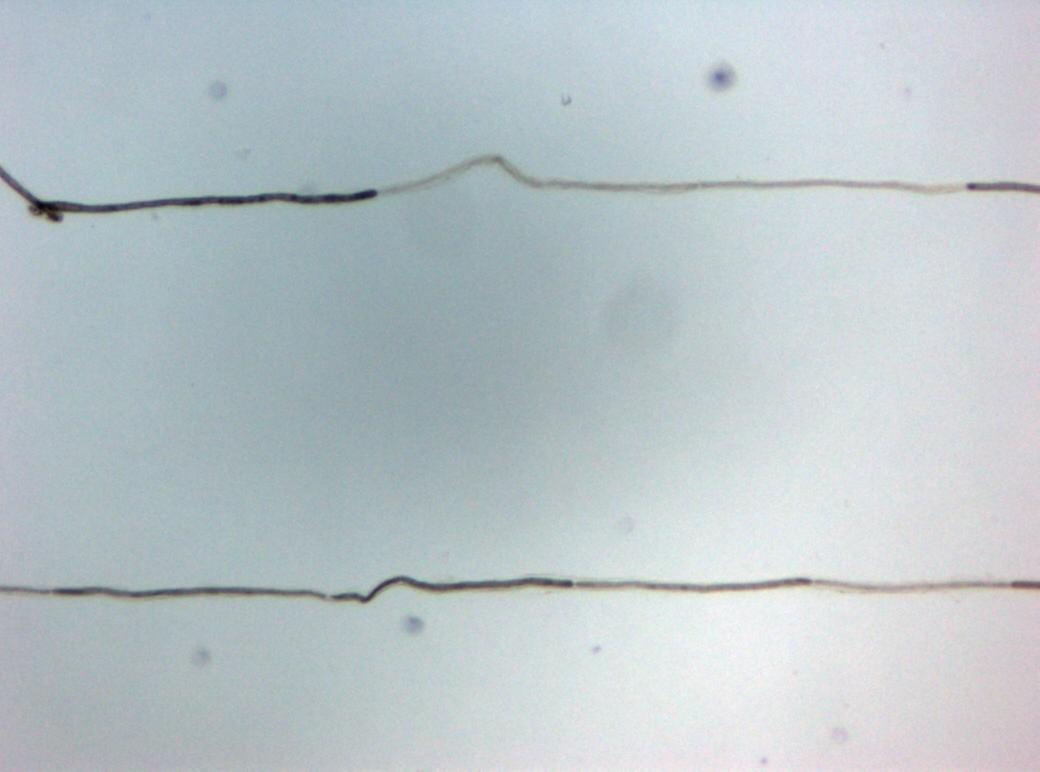
 Segmentaler Markscheidenverlust im Einzelfaserzupfpräparat
Segmentaler Markscheidenverlust im Einzelfaserzupfpräparat
Auch wenn die Durchführung einer Nervenbiopsie bei Erfüllung der übrigen diagnostischen Kriterien zur klinischen Diagnosesicherung nicht unbedingt erforderlich ist, wird meist eine Biopsie des Nervus suralis zur differentialdiagnostischen Sicherung der Diagnose herangezogen.[1] Im Kontext klinischer Studien ist die Durchführung einer Nervenbiopsie obligat (Research Criteria der American Academy of Neurology).[2]
Neben dem Nachweis einer demyelinisierenden entzündlichen Neuropathie im Semidünnschnitt kommt dem Nachweis einer segmentalen Demyelinisierung im Einzelfaserzupfpräparat eine besondere Bedeutung zu.
Therapiemöglichkeiten
Die Behandlung von CIDP erfordert eine besondere Vorgehensweise, da Patienten unterschiedlich darauf ansprechen. Da CIDP einen fortlaufenden (progredienten), wenn auch zuweilen aussetzenden Verlauf zeigt, ist meist eine länger dauernde medikamentöse Therapie erforderlich.
Anders als beim GBS zeigen Kortikosteroide bei der CIDP eine positive Wirkung. Sie verringern die Expression pro-inflammatorischer Cytokine und hemmen die T-Zellen-Entstehung (Proliferation). Auch die Plasmapherese und die intravenöse Immunglobulingabe stellen Therapie-Optionen dar. Allerdings muss die Therapie zyklisch alle 1-3 Monate wiederholt werden. Ungefähr zwei Drittel der Patienten zeigt einen positiven Therapieerfolg.
Nach Plasmapherese können sich die Symptome nach initialer kurzfristiger Besserung verschlechtern. Aufgrund der Nebenwirkungen einer langdauernden Steroid-Therapie werden additiv zur Dosis-Einsparung immunsuppressive Substanzen gegeben.
Dazu zählen unter anderem Azathioprin, Cyclophosphamid, Cyclosporin, Methotrexat, Mycophenolat-Mofetil, Rituximab und Interferon ß 1-a.
Die CIDP-Variante Lewis-Sumner-Syndrom spricht gut auf intravenöse Immunglobulingabe an, weniger gut auf Prednisolon.
Die CIDP-Variante mit zusätzlicher IgM-Gammopathie und MAGAK spricht kaum auf Prednisolon an. Unter intravenöser Immunglobulingabe kommt es zu inkomplettem Ansprechen.
Prognose
Das Erkrankungsalter scheint Einfluss zu haben auf den Verlauf. Patienten von weniger als 20 Jahren entwickeln häufig eine motorisch betonte Neuropathie mit subakuter Progression, relapsierend remittierendem Verlauf und guter Rückbildung. Patienten über 45 Jahren zeigen oft eine chronisch-progrediente sensomotorische Neuropathie mit verbleibenden neurologischen Defiziten.
Quellen
- GBS-Initiative (PDF) (798 kB)
Einzelnachweise
- ↑ Köller et al.: Akute und chronisch entzündliche Neuropathien - Diagnostik. Dtsch Med Wochenschr. 2003 Jun 13;128(24):1357-60. PMID 12802746
- ↑ Research criteria for diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP). Report from an Ad Hoc Subcommittee of the American Academy of Neurology AIDS Task Force. Neurology. 1991;41(5):617-8. PMID 2027473

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Autoimmunerkrankung
- Polyneuropathisches Syndrom
Wikimedia Foundation.