- Allylspannung
-
 Allyl-System
Allyl-System
Die Allylspannung (eigentlich 1,3-Allylspannung oder englisch 1,3-Allylstrain) ist ein Begriff aus der organischen Chemie und wurde 1968 von Francis Johnson definiert.[1] Man versteht darunter die Wechselwirkung zwischen den 1- und 3-Substituenten (R1 und R3) eines Allyl-Systems. Die ebenfalls existierende 1,2-Allylspannung ist dagegen von untergeordneter Bedeutung.[2]
Inhaltsverzeichnis
Beschreibung
Ein Allylsystem ist eine reaktionsfreudige funktionelle Gruppe in einem organischen Molekül. Sie ist gekennzeichnet durch eine Doppelbindung zwischen zwei Kohlenstoffatomen mit einer anschließenden Einfachbindung zu einem weiteren Kohlenstoffatom. Das einfachste Molekül mit einer solchen Gruppe ist das Propen (C3H6). Die Methylgruppe, die der Doppelbindung benachbart ist, ist in diesem Molekül frei drehbar, das heißt, es macht für den Energiegehalt des Moleküls keinen Unterschied, wie die Methylgruppe sich räumlich anordnet. Es gibt keine verschiedenen Konformationen, lediglich die für die Drehung der Wasserstoffatome gemessene Rotationsbarriere von 12,6 kJ/mol (siehe auch Konformer).
Trägt das der Doppelbindung benachbarte Kohlenstoffatom jedoch statt Wasserstoffatomen verschiedene Substituenten unterschiedlicher Größe, so ist diejenige Konformation am stabilsten, in welcher der kleinste Substituent in der Doppelbindungsebene liegt. Denn dieser Substituent tritt mit den π-Orbitalen der benachbarten Doppelbindung in Wechselwirkung, wodurch der Energiegehalt des Moleküls steigt. Diese erhöhte Energie bezeichnet man als Allylspannung. Weil jedes Molekül einen Zustand möglichst geringen Energiegehalts anstrebt, dreht sich die Einfachbindung zwischen den Kohlenstoffatomen im Molekül nun so, dass derjenige Substituent in diese Wechselwirkung tritt, mit dem die Allylspannung am geringsten ist. Sie ist damit eine sterische Hinderung bei (Z)-Alkenen. Außerdem können noch elektronische Effekte, wie zum Beispiel Hyperkonjugation, auftreten. In diesem Fall kann der sterisch kleinste Rest auch um ± 30° aus der Doppelbindungsebene gedreht sein, damit eine Wechselwirkung der σ–Bindung des Substituenten mit dem HOMO bzw. LUMO der π–Bindung ermöglicht wird.
Berechnungen ergaben dafür am Beispiel von sehr einfachen Allylsystemen folgende relativen Energieunterschiede:
Die 1,3-Allylspannung ist vergleichbar mit der 1,3-di-axialen Wechselwirkung an cyclischen Aliphaten und den Felkin-Anh-Regeln für Carbonylsysteme.[2]
Auswirkungen
Dieser Effekt lässt sich in der stereoselektiven Synthese ausnutzen.
So wird zum Beispiel bei der Hydroborierung, bei der ein neuer Substituent an die Doppelbindung addiert wird, dieser von der Seite her addiert, zu dessen Seite auch der kleinere Substituent am Vinylkohlenstoff steht.
Trägt einer der Reste R3 oder R4 eine Funktionalität, die für ein Reagenz, welches die C=C-Bindung angreift, dirigierend wirkt, so spricht man von einem aktiven Volumen, da in einer Reaktion der Angriff aus diesem Volumen (von dieser Seite) bevorzugt wird, durch Koordination an die Funktionalität, und dadurch eine stereochemische Information von der 1-Position auf die 3-Position übertragen wird.
Trägt einer der Reste R3 oder R4 keine koordinierend wirkende Funktionalität, sondern wirkt aufgrund seines sterischen Anspruches für ein Reagenz, welches die C=C-Bindung angreift, eher abschirmend, so spricht man von einem inerten Volumen, da in einer Reaktion der Angriff aus diesem Volumen (von dieser Seite) benachteiligt ist. Dadurch wird eine Stereoinformation von der 1-Position auf die 3-Position übertragen.
Beispiele
Durch die voluminöseren Reste erhöht sich der sterische Anspruch und die Diastereoselektivität steigt, da der Angriff nur noch von vorne (beta-Seite) erfolgen kann.
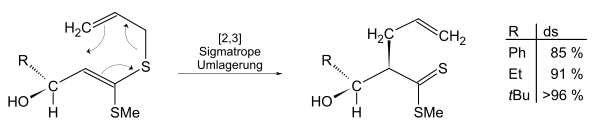
- Inert-Volumen am Beispiel einer [3,3]-sigmatropen Umlagerung.
meta-Chlorperbenzoesäure (mCPBA) wird von dem benzylgeschützten Alkohol sowie dem freien Alkohol koordiniert und greift das Molekül selektiv von hinten (alpha-Seite) unter Bildung des Epoxids an.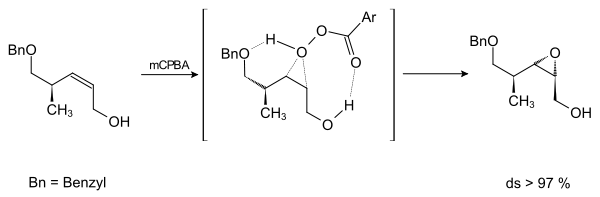
- Aktives Volumen am Beispiel einer mCPBA-Oxidation
Weitere Beispiele für stereoselektive Synthesen mit Hilfe der Allylspannung sind Cycloadditionen, Iodlactonisierungen und Epoxidierungen.[3][4]Literatur
- R. W. Hoffmann, Chemical Reviews 1989, 89, 1841–1860
- L. F. Tietze, Liebigs Annalen 1996, 1576–1579
Einzelnachweise





Wikimedia Foundation.