- PKD1
-

PKD1 (engl. Abk. für polycystic kidney disease 1 (autosomal dominant) = „polyzystische Nierenerkrankung 1 (autosomal-dominant)“) ist ein Gen, das sowohl beim Menschen als auch anderen Säugetier-Arten, im Genom enthalten ist.
Inhaltsverzeichnis
Funktion
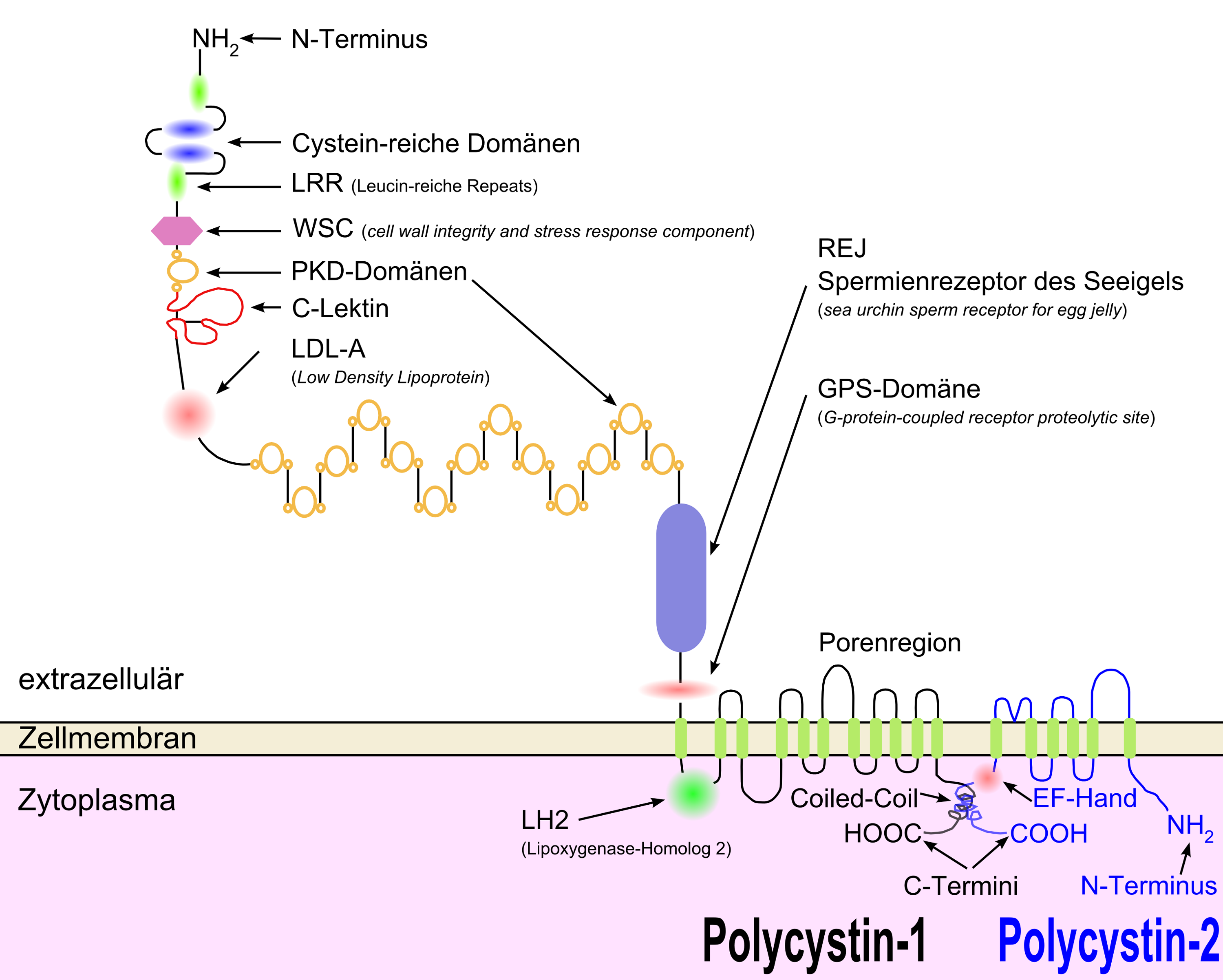
Das PKD1-Gen kodiert das Glycoprotein Polycystin-1. Polycystin ist ein multifunktionales Protein, das unter anderem bei der Reifung von Epithelzellen und bei der Aufrechterhaltung der renalen epithelialen Differenzierung, wie auch bei der Organisation der Struktur der Nephrone im frühen fetalen Stadium, eine wichtige Rolle spielt. Mutationen in PKD1 können autosomal-dominant vererbt werden und führen dann zur autosomal-dominanten Zystenniere (ADPKD) – der häufigsten lebensbedrohlichen Erbkrankheit beim Menschen. Bei Katzen führt dies zur polyzystischen Nierenerkrankung, die vor allem bei Perserkatzen auftritt.
Genetik
 Gerendertes Strukturmodell von Polycystin 1, das aus PKD1 kodiert wird
Gerendertes Strukturmodell von Polycystin 1, das aus PKD1 kodiert wird
Das PKD1-Gen befindet sich beim Menschen auf Chromosom 16 Genlocus p13.3. PKD1 ist eng mit dem Locus des Alpha-Globins, sowie dem PGP-Marker (Phosphoglycolatphosphatase) gekoppelt.[2] Diese Region, in welcher PKD1 liegt, umfasst 750 kb und ist reich an CpG-Dinukleotiden.[3] Das PKD1-Gen selbst umfasst 52 kb und enthält 50 Exons.[4] Von dem Gen sind verschiedene Spleiß-Varianten bekannt, die verschiedene Isoformen kodieren. Sechs Pseudogene von PKD1 auf Chromosom 16 wurden bisher beschrieben, die eine Ähnlichkeit von etwa 95 bis 97 % zu PKD1 aufweisen.[4] Diese Bereiche im Genom werden zwar transkribiert, aber durch das Fehlen eines geeigneten Startcodons nicht translatiert, da die mRNA so nicht an die Ribosomenuntereinheiten binden kann. Die hohe Zahl an Pseudogenen deutet auf eine mangelhafte Stabilität dieser Region auf Chromosom 16 hin.[5][6]
Mutationen
In einer 2001 veröffentlichten Studie wurde der gesamte kodierende Bereich von PKD1 auf potenzielle krankheitsauslösende Mutationen hin untersucht. Dabei wurden 69 Mutationen und 32 Polymorphismen entdeckt, die sich recht gleichmäßig über das gesamte Gen verteilen. Die Tatsache, dass nur drei der Mutationen auch auf den homologen Genen zu finden sind, wird dahingehend gedeutet, dass Genkonversionen nicht die Hauptursache für die hohe Mutationsrate von PKD1 sein können. 32% der in dieser Studie gefundenen Mutationen sind Nonsense-Mutationen und 29,6 % Deletionen und Insertionen. Mutationen an Spleißstellen haben einen Anteil von 6,2 %. Aus den vorliegenden Daten wurde eine Mutationsrate von 1,8×10-5 pro Generation errechnet. Dies stellt für ein menschliches Gen einen ungewöhnlich hohen Wert dar.[7][6]
Einzelnachweise
- ↑ C. Stayner und J. Zhou: Polycystin channels and kidney disease. In: Trends in Pharmacological Sciences 22, 2001, S. 543–546. PMID 11698076.
- ↑ M. L. Watson u. a.: Studies of genetic linkage between adult polycystic kidney disease and three markers on chromosome 16. In: J. Med. Genet. 24, 1987, S. 457–461. PMID 2821260.
- ↑ G. G. Germino u. a.: The gene for autosomal dominant polycystic kidney disease lies in a 750-kb CpG-rich region. In: Genomics 13, 1992, S. 144–151. PMID 1577479.
- ↑ a b J. Hughes u. a.: The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. In: Nature Genetics 10, 1995, S. 151–160. PMID 7663510.
- ↑ B. J. Loftus u. a.: Genome Duplications and Other Features in 12 Mb of DNA Sequence from Human Chromosome 16p and 16q. In: Genomics 60, 1999, S. 295–308. PMID 10493829.
- ↑ a b E. C. Kappe: Molekularbiologische Untersuchungen am PKD1-Gen der Katze. Dissertation, Justus-Liebig-Universität Giessen, 2008.
- ↑ S. Rossetti u. a.: Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. In: Am. J. Hum. Genet. 68, 2001, S. 46–63. PMID 11115377.
Weblinks
Wikimedia Foundation.