- Trost asymmetrische allylische Alkylierung
-
Die Trostsche asymmetrische allylische Alkylierung, oder einfach Asymmetrische allylische Alkylierung (AAA), ist eine Namensreaktion aus dem Bereich der Organischen Chemie. Sie dient meist der Einführung von Alkoholen, Aminen oder Derivaten von Phenol. Als Substrate werden Verbindungen benötigt, die eine Abgangsgruppe in allylischer Position besitzen. Die eingesetzten Katalysatoren sind meist Palladiumkomplexe. Die Reaktion ist benannt nach ihrem Entwickler, dem amerikanischen Chemiker Barry M. Trost.[1][2][3][4]
Die Reaktion wurde unter anderem als ein enantioselektiver Schlüsselschritt bei der Totalsynthese von Galantamin und Morphin eingesetzt.[5]
Inhaltsverzeichnis
Reaktionsmechanismus
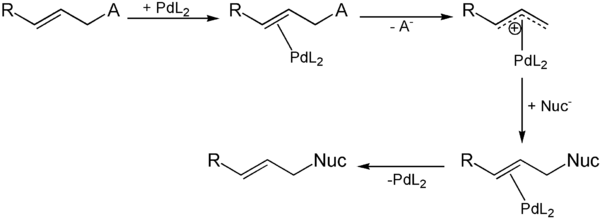
Zunächst bildet das eingesetzte Alken mit dem Palladiumkatalysator einen π-Komplex. Unter Abspaltung der Abgangsgruppe wird dann ein kationischer Komplex gebildet. Das Nucleophil greift nun an der Position, die vorher die Abgangsgruppe getragen hat, an unter Bildung eines zweiten π-Komplexes. Durch Abspaltung des Katalysators wird das gewünschte Produkt freigesetzt.
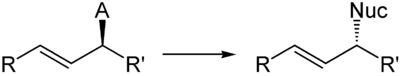
Bei Verwendung von chiralen Substraten tritt am Stereozentrum Inversion ein.
Die Reaktion wird im basischen Milieu geführt. Als Basen können Alkylamine wie Triethylamin eingesetzt werden.
Edukte und Katalysatoren
 Trost-Ligand
Trost-LigandDer benötigte Palladiumkatalysator der Oxidationsstufe 0 wird in situ erzeugt. Als Liganden eignen sich Phosphinliganden, beispielsweise der Trost-Ligand.
Als Abgangsgruppen eignen sich elektronenarme und somit saure Carbonsäureanionen, beispielsweise Acetate oder Trichloracetate. Chirale Substrate sind aus den entsprechenden chiralen Alkoholen zugänglich.
Zur Einführung von Hydroxylgruppen kann Wasser als Nucleophil verwendet werden, Ether können unter Einsatz von Alkoholen erhalten werden. Zur Einführung von Aminen eignen sich Phthalimidsalze. Aus dem erhaltenen Produkt kann in einem weiteren Reaktionsschritt das gewünschte Amin freigesetzt werden.
Modifikationen
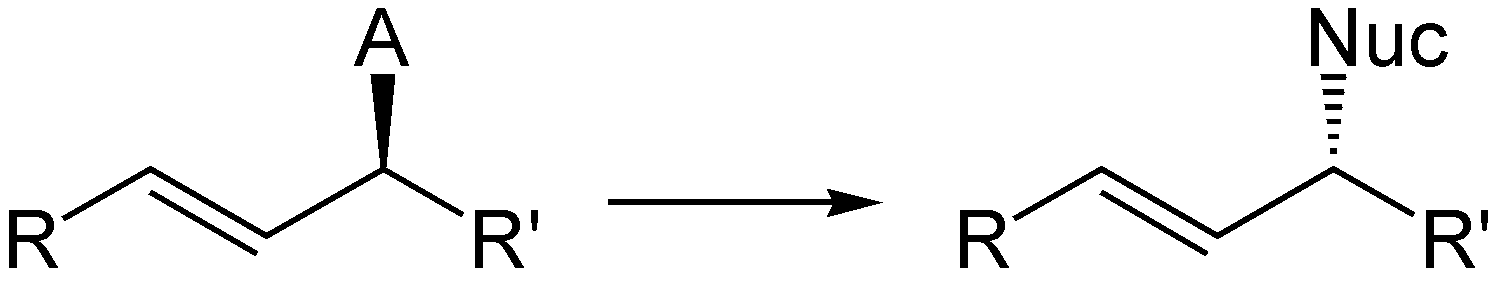
Die komplementäre Reaktion wurde von Günter Helmchen entwickelt.[6][7][8] Unter Verwendung von Iridiumkatalysatoren mit chiralen Phosphoramiditliganden können aus achiralen internen Alkenen chirale terminale Alkene synthetisiert werden. Das Nucleophil greift hierbei im Gegensatz zur Trost-Allylierung nicht an der Position an, die die Abgangsgruppe getragen hat, was auf die unterschiedlichen elektronischen Eigenschaften von Palladium und Iridium zurückzuführen ist. Als Abgangsgruppen können ebenfalls Carbonsäureanionen verwendet werden, meistens werden jedoch organische Carbonate eingesetzt.
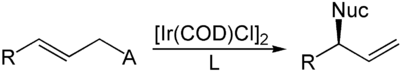
Quellen
- ↑ B. M. Trost, T. J. Fullerton: New synthetic reactions. Allylic alkylation., in: J. Am. Chem. Soc. 1973, 95, 292–294.
- ↑ B. M. Trost, T. J. Dietsch: New synthetic reactions. Asymmetric induction in allylic alkylations., in: J. Am. Chem. Soc. 1973, 95, 8200–8201.
- ↑ B. M. Trost, P. E. Strege: Asymmetric induction in catalytic allylic alkylation., in: J. Am. Chem. Soc. 1977, 99, 1649–1651.
- ↑ B. M. Trost, M. L. Crawley: Asymmetric Transition-Metal-Catalyzed Allylic Alkylations:Applications in Total Synthesis, in: Chem. Rev. 2003, 103, 2921–2944.
- ↑ B. M. Trost, W. Tang, F. D. Toste: Divergent Enantioselective Synthesis of (−)-Galanthamine and (−)-Morphine., in: J. Am. Chem. Soc. 2005, 127, 14785–14803.
- ↑ J. P. Janssen, G. Helmchen: First Enantioselective Alkylations of Monosubstituted Allylic Acetates Catalyzed by Chiral Iridium Complexes, in: Tetrahedron Lett. 1997, 109, 8025–8026.
- ↑ B. Bartels, G. Helmchen: Ir-catalysed allylic substitution: mechanistic aspects and asymmetric synthesis with phosphorus amidites as ligands, in: Chem. Commun. 1999, 741–742.
- ↑ G. Lipowsky, N. Miller, G. Helmchen: Regio- und enantioselektive Iridium-katalysierte allylische Alkylierung mit in situ aktivierten P,C-Chelatkomplexen, in: Angew. Chem. 2004, 116, 4695–4698.



Wikimedia Foundation.