- Chiralität (Chemie)
-
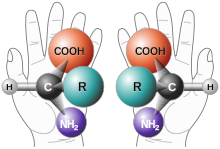 Die beiden Enantiomere eines chiralen Moleküls finden ihre Entsprechung im Alltag in der rechten und linken Hand.
Die beiden Enantiomere eines chiralen Moleküls finden ihre Entsprechung im Alltag in der rechten und linken Hand.
In der Chemie bezeichnet die Chiralität (griechisches Kunstwort, die Händigkeit, abgeleitet vom Wortstamm χειρ~, ch[e]ir~ – hand~), in der Kristallographie auch Enantiomorphie genannt, die räumliche Anordnung von Atomen, bei denen bestimmte Symmetrieoperationen, zum Beispiel die Spiegelung an einer Molekülebene, nicht zu einer Selbstabbildung führen. Hierbei können sowohl einzelne oder mehrere Atome in einem Molekül eines oder mehrere stereogene Zentren darstellen als auch die gesamte Molekülgestalt die Chiralität ausmachen. Moleküle mit dieser Eigenschaft werden dabei chiral, Moleküle ohne diese Eigenschaft achiral genannt.
Gängige Beispiele aus dem Alltagsleben sind rechte und linke Hand oder rechts- bzw. linksgewundene Schneckenhäuser.
Allgemein ist ein Objekt genau dann chiral, wenn es keine Drehspiegelachse besitzt. Andere Symmetrieelemente können aber durchaus vorhanden sein, d. h. ein chirales Objekt ist nicht zwangsläufig asymmetrisch.
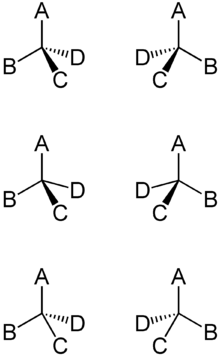 Spiegelbildlich gezeichnete Enantiomerenpaare in (von oben nach unten) drei verschiedenen (gleichwertigen) Visualisierungsformen.
Spiegelbildlich gezeichnete Enantiomerenpaare in (von oben nach unten) drei verschiedenen (gleichwertigen) Visualisierungsformen.Inhaltsverzeichnis
Chemie allgemein
Chiralität beruht meist auf der unterschiedlichen räumlichen Anordnung von Atomen und Atomgruppen um eines oder mehrere Stereozentren. So stellen z. B. Kohlenstoffatome mit vier verschiedenen Substituenten ein stereogenes Zentrum oder Stereozentrum dar, bei dem zwei verschiedene räumliche Anordnungen möglich sind. Neben Kohlenstoff können auch andere Atome wie zum Beispiel Phosphor Stereozentren ausbilden. Entscheidend ist hierbei, dass die Substituenten ihre relative Lage zueinander nicht ändern können, was im Falle des Phosphors durch eine ausreichend große Inversionsbarriere gewährleistet ist. Stickstoff kann meist nur in gespannten Systemen als Stereozentrum fungieren, da Stickstoff sonst in hoher Frequenz oszilliert und somit ständig invertiert. Am chiralen Stickstoffzentrum gilt das freie Elektronenpaar als vierter Substituent. Dieser besitzt die niedrigste Priorität aller Substituenten.
Moleküle, deren Bild und Spiegelbild sich nicht zur Deckung bringen lassen, sind also chiral. Die beiden somit unterscheidbaren spiegelbildlichen Formen eines solchen Moleküls werden als Enantiomere bezeichnet. Die Enantiomere können durch ihre unterschiedliche optische Aktivität unterschieden werden. Eine Mischung mit gleichen Anteilen beider Enantiomere wird Racemat oder racemisches Gemisch genannt.
Im einfachsten Fall liegt in der organischen Chemie Chiralität dann vor, wenn in einem Molekül ein Kohlenstoffatom vier verschiedene Substituenten trägt. Dieses Kohlenstoffatom wird als Stereozentrum (manchmal auch veraltet als Chiralitätszentrum oder asymmetrisches Kohlenstoffatom) bezeichnet. Grundlegende Überlegungen und Messungen zur Chiralität entsprechend substituierter Kohlenstoffverbindungen gehen auf Jacobus Henricus van't Hoff und (später) Paul Walden zurück. Die räumliche Anordnung der Substituenten an einem Stereozentrum wird nach den durch R. S. Cahn, C. K. Ingold und V. Prelog festgesetzten Regeln (CIP-Regeln) mit (R) oder (S) [(R) für rectus, lateinisch „rechts“, und (S) für sinister, lat. „links“] bezeichnet. Liegen mehrere Stereozentren vor, erhöht sich auch die Anzahl möglicher verschiedener Verbindungen. Mit n Stereozentren ergeben sich 2n verschiedene Verbindungen, abzüglich möglicher meso-Verbindungen (s. u.). Die Stereozentren werden dann jeweils einzeln nach den CIP-Regeln mit (R) oder (S) bezeichnet. Unterscheiden sich zwei Verbindungen in einem oder mehreren, nicht aber in allen Stereozentren, so spricht man von Diastereomeren. Wenn ein Molekül mehrere Stereozentren aufweist, diese aber durch eine Spiegelung an einer Ebene ineinander überführt werden können, so ist das gesamte Molekül achiral. Man spricht in diesem Fall von meso-Verbindungen (z. B. meso-Weinsäure).
Eine ältere Konvention zur Benennung von Enantiomeren, die heute noch für Zucker und teilweise auch für Aminosäuren angewandt wird, ist die D- und L-Nomenklatur des Nobelpreisträgers Emil Fischer (Fischer-Projektion).
Neben dieser auf Stereozentren zurückgeführten Chiralität (zentrale Chiralität) unterscheidet man axiale, planare und helicale Chiralität, um die zugrundeliegenden Strukturelemente näher zu beschreiben. Axiale Chiralität tritt z. B. bei Biphenylen wie BINAP auf, die so in den ortho-Positionen substituiert sind, dass die freie Drehbarkeit der Aromaten um die C-C-Einfachbindung stark gehindert ist. Hieraus ergeben sich dann zwei spiegelbildliche Isomere. Beispiele für planare Chiralität sind E-Cycloocten oder bestimmte Sandwich-Komplexe oder substituierte Cyclophane. Unter helicaler Chiralität versteht man den unterschiedlichen Drehsinn helicaler Verbindungen. Helicale Chiralität tritt z. B. bei Helicenen auf.
Allen chiralen Verbindungen ist die Abwesenheit einer Drehspiegelachse gemeinsam. Wie sich aus der Gruppentheorie beweisen lässt, ist diese Abwesenheit einer Drehspiegelachse die notwendige und ausreichende Bedingung dafür, dass ein Molekül in Enantiomeren auftritt. Eine Drehspiegelachse Sn ist ein Symmetrieelement. Hierbei dreht man das Molekül zuerst um 360/n Grad um eine Achse und spiegelt es anschließend an der Ebene, die senkrecht zu dieser Achse liegt. Ist das Produkt dieser Operation identisch mit der Ausgangsverbindung, hat man eine Drehspiegelachse gefunden.
Des Weiteren existieren auch noch die Begriffe Pseudochiralität und Prochiralität. Zwei der Substituenten an einem pseudochiralen (=pseudoasymmetrischen) Zentrum unterscheiden sich nur durch ihre Konfiguration, sind also enantiomorph. Dadurch liegen sie auf einer Spiegelebene des Moleküls (sofern keine weiteren stereogenen Zentren vorhanden sind), es ist damit eine achirale, sogenannte meso-Verbindung. (Teilweise wird der Begriff auch dann benutzt, wenn beide Substituenten gleich konfiguriert sind und das Molekül damit chiral ist, zum Beispiel in [1]). Im Vergleich zur R/S-Nomenklatur (CIP-Nomenklatur) werden pseudochirale Zentren mit r und s bezeichnet, wobei der (R)-konfigurierte Substituent die höhere Priorität erhält. Prochirale Gruppen sind solche Funktionen, die durch eine Addition an ihr selbst in ein Stereozentrum überführt werden können. Als gutes Beispiel dienen hier unsymmetrische Ketone, die beispielsweise durch Hydrierung in chirale Alkohole überführt werden können. Man unterscheidet hierbei den Angriff von der re- oder si-Seite.
Chiralität tritt auch in anorganischen Stoffen auf. So besitzt der Quarz zwei enantiomorphe Formen, die sich auf links- oder rechtsgängige Schrauben zurückführen lassen. Dies ist ebenfalls ein Beispiel für helicale Chiralität. In der Kristallographie gibt es insgesamt elf enantiomorphe Punktgruppen.
Die absolute Konfiguration einer chiralen Substanz kann nicht aus dem Drehsinn von polarisiertem Licht beim Passieren einer Standardlösung erschlossen werden, sondern muss entweder durch chemische Analogieschlüsse (zum Beispiel durch Abbau der zu bestimmenden Substanz zu einer bekannten Verbindung), durch Röntgenkristallographie oder durch Verwendung chiraler Shift-Reagenzien in der NMR-Spektroskopie erfolgen. Erst nach einem solchen Nachweis, kann entschieden werden, ob eine Verbindung (R)- oder (S)-Konfiguration besitzt. Die Zuordnung der Konfigurationen für Aminosäuren und Kohlenhydrate, von denen Anfangs nur die relativen Konfigurationen zueinander bekannt waren, erfolgte zunächst willkürlich. Sehr viel später (in den 1950er Jahren) haben röntgenkristallographische Untersuchungen ergeben, dass die gewählte Zuordnung zufälligerweise (Hinweis: Wahrscheinlichkeit dafür betrug 50 %) den tatsächlichen Verhältnissen entspricht. Siehe auch bei Circulardichroismus.
Biochemie
Das Konzept der Chiralität spielt auch in der Biologie, insbesondere in der Biochemie, eine fundamentale Rolle. In allen Naturstoffklassen ist jeweils ein Enantiomer bevorzugt, bzw. ausschließlich vorhanden. So findet man in der Natur z. B. ausschließlich D-Glucose und keine L-Glucose. (Es gibt aber durchaus L-Zucker, und sogar Zucker die sowohl in der D- als auch in der L-Form vorkommen, allerdings in jeweils völlig unterschiedlichen Zusammenhängen, siehe Monosaccharide). Auf dieses in der Natur vorhandene Reservoir enantiomerenreiner Substanzen, den Chiral Pool, sind viele stereoselektiven Synthesen und enantiomerenreine synthetische Verbindungen in direkter oder indirekter Form, etwa durch stereoselektive Katalysatoren, zurückzuführen.
Biochemische Reaktionen werden durch Enzyme katalysiert. Da es sich bei Enzymen um chirale Makromoleküle handelt, sind sie in der Lage, eine Reaktion enantioselektiv zu steuern. Dies geschieht durch einen diastereoselektiven (!) Mechanismus, wobei von den beiden enantiomeren Übergangszuständen des Substrates derjenige bevorzugt wird, dessen Energie geringer ist, der also vom aktiven Zentrum stabilisiert wird. Dadurch können aus prochiralen und achiralen Edukten chirale Produkte synthetisiert werden. Auf diese Weise setzt sich die Bevorzugung eines Enantiomers und damit die Chiralität in der gesamten Biochemie und Physiologie fort. Chiralität ist auch die Voraussetzung für geordnete Tertiärstrukturen wie z. B. einer α-Helix, die nur aus enantiomerenreinen Aminosäuren aufgebaut werden kann (in der Natur L-Aminosäuren). Enantiomere chiraler Moleküle zeigen somit in der Regel unterschiedliche physiologische Wirkungen, sie haben einen unterschiedlichen Geschmack, Geruch, eine unterschiedliche Toxizität und eine unterschiedliche pharmakologische Wirkung als Arzneistoff.[2][3] (Bei dem bekanntesten Fall Contergan®/Thalidomid ist die unterschiedliche Wirkung der Enantiomere allerdings nicht genau abzugrenzen, da es in vivo zu einer Racemisierung kommt und deshalb die differierende physiologische Wirkung der Enantiomeren 'eingeebnet' und damit irrelevant wird).
Genau genommen wäre aus diesem Grund eine „racemische Biologie“ im Sinne eines 1:1-Gemisches aller Enantiomeren gar nicht möglich, da für die spiegelverkehrten Moleküle ein kompletter eigener ebenfalls spiegelverkehrter Syntheseapparat notwendig wäre. Die in manchen bakteriellen Zellwänden proteinogen gebunden vorkommenden raren D-Aminosäuren werden über einen Sekundärmetabolismus synthetisiert und verhindern z. B. den Abbau durch Proteasen.
Es ist bis heute nicht geklärt, ob die angetroffene Bevorzugung eines bestimmten Enantiomers von Biomolekülen (z. B. besitzen praktisch alle natürlich vorkommenden Aminosäuren L- und nicht D-Konfiguration) sich auf eine zufällige Selektion am Beginn der Evolutionskette begründet, die sich dann selbst verstärkt hat, oder ob es fundamentale Gründe für die Bevorzugung dieser Konfiguration gibt. Aufgrund der schon oben erwähnten Paritätsverletzung bei der Schwachen Wechselwirkung ist nämlich der Energiegehalt zweier Enantiomere nicht exakt gleich. Der Energieunterschied ist jedoch so gering, dass mit Recht die Frage gestellt werden kann, ob er überhaupt bedeutsam ist.
Biokatalyse
Bei biokatalytischen Transformationen wird ausgenutzt, dass bei Umsetzungen mit Enzymen als Biokatalysator zumeist ein Enantiomer im Überschuss entsteht, beziehungsweise wenn ein Racemat als Ausgangsmaterial vorgelegt wird, vom Enzym ein Enantiomer bevorzugt umgesetzt wird. So kann ausgehend von einem racemischen Ester die Estergruppe eines Enantiomers des Esters unter stereoselektivem Einfluss des Enzyms Pankreaslipase hydrolysiert werden, während das andere Enantiomer des Esters unverändert bleibt. Die enantiomerenreine Carbonsäure kann dann leicht von dem auch weitestgehend enantiomerenreinen Ester mit üblichen Trennverfahren (Kristallisation, Chromatographie etc.) abgetrennt werden. Das Enzym Aspartase kann die enantioselektive Addition von Ammoniak an die C=C-Doppelbindung von Fumarsäure katalysieren, es entsteht gezielt (S)-Asparaginsäure.
Siehe auch
Literatur
- Pedro Cintas: Ursprünge und Entwicklung der Begriffe Chiralität und Händigkeit in der chemischen Sprache. Angewandte Chemie 119(22), S. 4090–4099 (2007), ISSN 0044-8249.
- Henri Brunner: Rechts oder Links, Wiley-VCH Verlag, Weinheim/Bergstasse, 1999, ISBN 3-527-29974-2.
- Uwe Meierhenrich: Amino Acids and the Asymmetry of Life, Springer-Verlag, Heidelberg, Berlin 2008. ISBN 978-3-540-76885-2.
Weblinks
- ChemgaPedia: Axiale Chiraliät, Helicale Chiralität, Planare Chiralität
Einzelnachweise
- ↑ Arnold Fr. Holleman, Egon Wiberg: Lehrbuch der Anorganischen Chemie. 101., verb. u. stark erw. A. Auflage. Gruyter, 1995, ISBN 3110126419, S. 747.
- ↑ E. J. Ariëns, Stereochemistry, a basis for sophisticated nonsense in pharmacokinetics and clinical pharmacology, European Journal of Clinical Pharmacology 26 (1984) 663-668.doi:10.1007/BF00541922
- ↑ Hisamichi Murakami: From Racemates to Single Enantiomers – Chiral Synthetic Drugs over the last 20 Years, Topics in Current Chemistry 269 (2007) 273−299, doi 10.1007/128_2006_072.

Wikimedia Foundation.