- Racematspaltung
-
Als Racematspaltung werden Verfahren zur Trennung von Racematen in ihre Enantiomere bezeichnet. Enantiomere von Wirkstoffen in Pharmaka oder Pflanzenschutzmitteln besitzen meist eine unterschiedliche biologische Aktivität. Die Racematspaltung wird unter anderem dazu eingesetzt, diese Enantiomere in möglichst reiner Form zu gewinnen. Allein der Umsatz von Arzneimitteln mit enantiomerenreinen Arzneistoffen betrug im Jahr 2000 etwa 150 Milliarden US-Dollar.[1]
Bis zur Entwicklung von asymmetrischen Synthesemethoden war die Racematspaltung die einzige Möglichkeit zur Gewinnung reiner Enantiomere aus racemischen Produkten, die bei chemischen Synthesen meist entstehen. Die Racematspaltung ist bis heute eine industriell häufig angewandte Methode zur Erzeugung enantiomerenreiner Stoffe.
Inhaltsverzeichnis
Geschichte
 Louis Pasteur
Louis Pasteur
Die erste Racematspaltung gelang Louis Pasteur im Jahr 1848 durch manuelles Aussortieren von enantiomeren Natrium-Ammonium-Tartrat-Kristallen, die sich makroskopisch wie Bild und Spiegelbild verhalten, unter dem Mikroskop[2]. Eine Voraussetzung dafür ist, dass das Racemat von sich aus durch spontane Spaltung Kristalle bildet, die nur eines der Enantiomere enthalten. Im Jahr 1857 gelang Pasteur die Racematspaltung durch Diastereomerentrennung. Durch Kombination einer racemischen Säure (oder Base) mit einer optisch aktiven Base (oder Säure) zu einem diastereomeren Salz und anschließender fraktionierter Kristallisation konnten die Enantiomere getrennt werden.[3] Pasteur fand noch eine dritte Methode der Racematspaltung. Bereits 1858 nutzte er erstmals das Verfahren der Racematspaltung durch Fermentation mittels des Schimmelpilzes Penicillium glaucum, den er auf racemischer Weinsäure als Nährstoff wachsen ließ. Während ein Enantiomer vom Pilz verstoffwechselt wurde, blieb das andere Enantiomer in der Lösung zurück.[4]
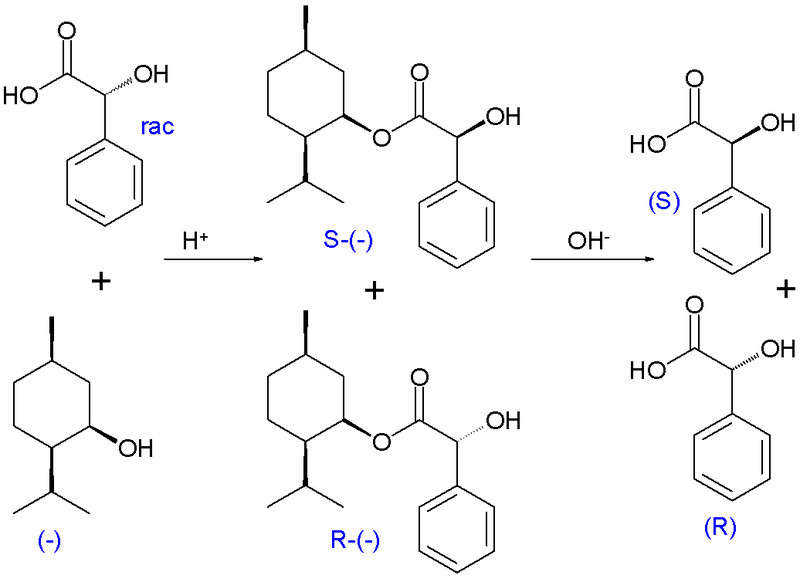
Marckwald und McKenzie berichteten 1899 über die erste kinetische Racematspaltung bei der Veresterung von racemischer Mandelsäure mit optisch aktiven (–)-Menthol. Bei dieser Reaktion zeigt das (R)-Enantiomer der Mandelsäure eine höhere Reaktionsgeschwindigkeit und im Reaktionsgemisch wird (S)-Mandelsäure anreichert.[5]
Die erste gaschromatografische Trennung von D,L-Aminosäuren-Racematgemischen in ihre Enantiomere mittels einer chiralen stationären Phase gelang 1966 Emanuel Gil-Av am Weizmann-Institut für Wissenschaften in Rehovot, Israel.[6]
Im Jahr 1975 meldete Merck ein Patent zur Herstellung von Methyldopa über Racematspaltung mittels Konglomeratkristallisation an.[7]
Im Jahr 2002 startete die Degussa (heute Evonik Degussa GmbH) die Produktion von enantiomerenreinen Aminosäuren mit dem Hydantoinase-Verfahren, das auf dynamischer kinetischer Racematspaltung basiert.[8]
Mechanische Trennverfahren
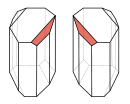 D- und L-Tartratkristalle, die sich wie Bild und Spiegelbild verhalten
D- und L-Tartratkristalle, die sich wie Bild und Spiegelbild verhaltenDie von Pasteur angewandte Methode der mechanischen Trennung eignet sich nur für Systeme, in denen die Enantiomere als enantiomerenreine Konglomerate kristallisieren. Außerdem können so nur relativ geringe Mengen getrennt werden.
Besser gelingt die Trennung von Racematen durch Animpfen übersättigter Racemat-Lösungen mit einer geringen Menge eines reinen Enantiomers des gleichen Racemates und anschließende fraktionierende Kristallisation.[9] Die Trennung von enantiomerenreinem Kristall und Lösung geschieht durch mechanische Verfahren wie Filtration oder Zentrifugation. Diese Methode wurde zwar im industriellen Maßstab eingesetzt, ist in ihrer Anwendungsbreite eingeschränkt, da viele Racemate auch racemisch und nicht als Konglomerat kristallisieren.
Trennverfahren über Diastereomerenbildung
Die in einem Racemat enthaltenen Enantiomere lassen sich durch Reaktion mit einem chiralen Reagenz oder Kontakt mit einer chiralen Phase in Diastereomere überführen, die unterschiedliche physikalische Eigenschaften besitzen. Durch die Unterschiede in den physikalischen Eigenschaften lassen sich die entstehenden Diastereomere mittels herkömmlicher Verfahren wie Chromatographie, Kristallisation oder Destillation trennen. Dabei muss die Bildung des Diastereomers nicht unbedingt über kovalente Bildung erfolgen, es kann sich dabei auch um diastereomere Übergangszustände, die etwa durch Wasserstoffbrückenbindung entstehen, wie etwa bei der chiralen Chromatographie handeln. Eine Voraussetzung für diese Trennmethode ist es, dass die Trennung schneller als Racemisierung erfolgt oder dass stabile Isomere vorliegen. Eine weitere Voraussetzung ist, dass etwa aus dem Chiral pool enantiomerenreine Verbindungen vorliegen, die zur Bildung der Diastereomeren eingesetzt werden können.
Chirale Chromatographie
Racemate lassen sich mit allen bekannten Chromatographiemethoden wie der Dünnschichtchromatographie, der HPLC, Säulenchromatografie oder der Gaschromatografie trennen. Letztere findet allerdings bevorzugt in der Analytik nicht-racemischer Enantiomerengemische Anwendung zur Bestimmung des Enantiomerenüberschusses.
Die chirale Phase kann dabei stationär sein oder im Falle der Flüssigkeitschromatographie auch das Eluent. Die im organisch-chemischen Labor übliche Methode ist das in Kontakt bringen mit chiralen Materialien. Chromatographisch wählt man dazu entweder die mobile Phase (Eluenz) oder die stationäre Phase optisch aktiv. Das führt zur unterschiedlichen Retention zweier Enantiomere. Auch eine dünnschichtchromatographische Enantiomerentrennung unter Verwendung einer enantioselektiven stationären Phase ist bekannt.[10]
Größere Probenmengen lassen sich über Säulenchromatographie trennen. Zur Herstellung der chiralen stationären Phase wird z. B. ein enantiomerenreines chirales Auxiliar wie (+)-Weinsäure auf einem Träger, wie z.B. Kieselgel fixiert. Die racemische Lösung wird mit einem herkömmlichen Eluent chromatographiert. Die einzelnen Enantiomere wechselwirken unterschiedlich stark mit der chiralen Matrix und verlassen die Säule bei verschiedenen Retentionszeiten.
Die Differenz der freien Standardbildungsenergie der diastereomeren Übergangszustände
- ΔS, RΔG≠ = RT ln α
(α: Trennfaktor)
bewirkt einen Unterschied der Retentionszeit.
Als chirale Phasen finden eine Reihe von Stoffen Anwendung. Die Art des chiralen Erkennens ist dabei je nach Art des Trägermaterials verschieden. Peptidphasen wechselwirken meist über Wasserstoffbrücken sowie Dipol-Dipol-Wechselwirkungen, während chirale Metallkomplexe über Komplexierung wechselwirken. Intensiv untersucht wurde die Gaschromatographie mittels Cyclodextrinderivaten.[11][12]
Kristallisation und Destillation
Die Racematspaltung mittels fraktionierter Kristallisation ist eine weit verbreitete Methode. Dabei werden diastereomere Salze durch Zugabe eines enantiomerenreinen Hilfsstoffes und anschließende Trennung durch fraktionierende Kristallisation unter Ausnutzung ihrer unterschiedlichen physikalischen Eigenschaften getrennt.[13] Weit verbreitet ist der Einsatz von Weinsäure oder Chinin als Komponente des Chiral pools.
Nicht oder schwer kristallisierbare Diastereomere können auch destillativ getrennt werden. Das Verfahren eignet sich etwa für racemische Säuren oder Alkoholgemische, die in die Ester überführt werden können.
Kinetische Racematspaltung
 Prinzip der kinetischen Racematspaltung
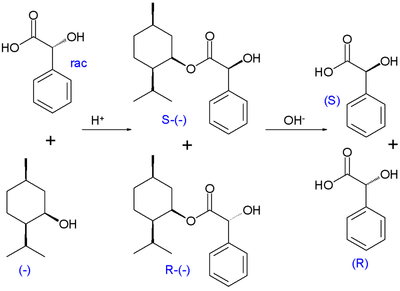
Prinzip der kinetischen RacematspaltungSind die Geschwindigkeitskonstanten der Überführung der Substratisomere SR und SS in die korrespondierenden Produkte PR und PS verschiedenen, so liegt eine kinetische Racematspaltung vor. Die Reaktion wird bei einem Umsatz von circa 50 % abgebrochen, da dann das schneller reagierende Enantiomer verbraucht ist. Im Reaktionsgemisch reichert sich das langsamer reagierende Enantiomer an. Die Komponenten SS und PR können mit herkömmlichen Methoden getrennt werden, PR kann nach erfolgter Trennung gegebenenfalls wieder in SR überführt werden.
Bei der kinetischen Racematspaltung nach Willi Marckwald und McKenzie wird racemische Mandelsäure mit optisch aktivem (−)-Menthol teilverestert. Das (R)-Enantiomer der Mandelsäure zeigt dabei eine höhere Reaktionsrate und in der Reaktionsmischung reichert sich die (S)-Mandelsäure an. Die angereicherte Fraktion kann abgetrennt werden, die (R)-Mandelsäure kann durch Hydrolyse wieder zurückgewonnen werden.
Im Gegensatz zur fermentativen Racematspaltung haben kinetische Racematspaltungen mittels Enzymen eine breite Anwendung gefunden.[14]
Dynamische kinetische Racematspaltung
 Prinzip der dynamischen kinetischen Racematspaltung
Prinzip der dynamischen kinetischen RacematspaltungDie Nachteile der kinetischen Racematspaltung wie die theoretisch begrenzte Ausbeute von 50% und die notwendige Aufarbeitung der Reaktionslösung lassen sich durch die dynamische kinetische Racematspaltung vermeiden. Durch die Racemisierung des langsamer reagierendes Enantiomers SS lassen sich Racemate quantitativ in Produkte mit hohem Enantiomerenüberschuss umwandeln. Die Ausbeute und der Enantiomerenüberschuss kann theoretisch 100% betragen.
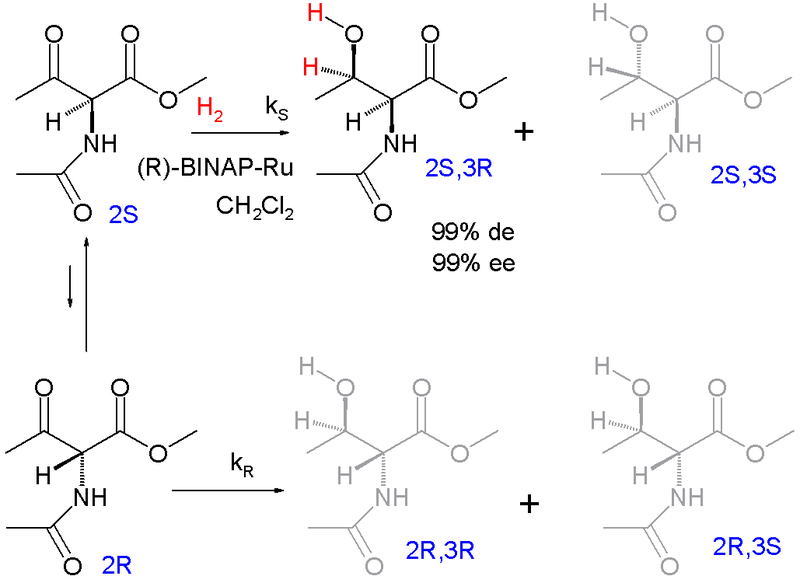
Eines der ersten Beispiele für die dynamische kinetische Racematspaltung ist die asymmetrische Hydrierung nach Ryoji Noyori (1989):[15]
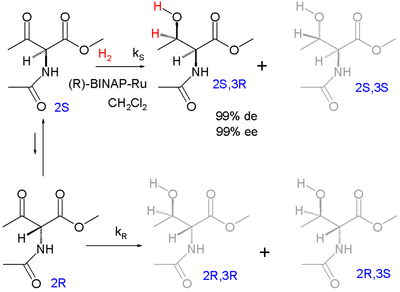
Die Enantiomere racemisieren über die Enol-Form. Das Zielprodukt ist das geschützte syn-Addukt l-Threonin (2S, 3R) mit 99% Diastereomerenüberschuss (mit Präferenz für das syn-Diastereomerenpaar und nicht das Anti-Paar) und 99% Enantiomerenüberschuss (Präferenz für das (3R)-Produkt innerhalb der syn-Paares).
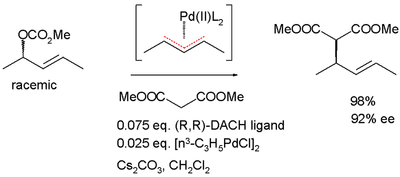
Die dynamische kinetische Racematspaltung kann auch über die Bildung eines prochiralen Übergangszustandes oder einer meso-Verbindung erfolgen. Ein Beispiel hierfür ist die allylische asymmetrische Alkylierung nach Barry Trost, die über einen η3-Palladium-Allylkomplex erfolgt.[16]
Ein Beispiel für die industrielle Anwendung der dynamischen kinetischen Racematspeltung ist das von der Degussa entwickelte Hydantoinase-Verfahren zur Darstellung von Aminosäuren mit hoher Enantiomerenreinheit. Als Substrate werden Hydantoine verwendet, die durch Zugabe von Racemase schnell racemisiert werden. Durch D-Hydantoinase wird das (R)-Hydantoin zur Carbamoyl-geschützten D-Aminosäure gespalten, die dann durch eine Carbamoylase anschließend entschützt wird.
Literatur
- G. Subramanian: Chiral Separation Techniques: A Practical Approach, 641 Seiten, Verlag Wiley-VCH Verlag GmbH & Co. KGaA (2006) ISBN 3-527-31509-8, ISBN 978-3-527-31509-3.
- W. A. König: The Practice of Enantiomer Separation by Capillary Gas Chromatography, 104 Seiten, Verlag Hüthig (1987); ISBN 3-7785-1324-9, ISBN 978-3-7785-1324-8.
- P. J. Walsh, M. C. Kozlowski: Fundamentals of Asymmetric Catalysis, 688 Seiten, Verlag Palgrave Macmillan (2008), ISBN 1-891389-54-8, ISBN 978-1-891389-54-2.
- A. Pandey: Enzyme Technology, 742 Seiten, Verlag Springer, Berlin (2006), ISBN 0-387-29294-2, ISBN 978-0-387-29294-6.
Einzelnachweise
- ↑ Bedeutung der Chiralität und Enantiomerentrennung - Methoden der Chiralitätserkennung, von V. Schurig, Institut für Organische Chemie, Universität Tübingen.
- ↑ Pasteur, M. L. (1848): Ann. Chim. Physik Bd. 24, S. 442.
- ↑ Pasteur, M. L. (1857): C. R. Hebd. Seance Acad. Sci., Bd. 45, S. 1032.
- ↑ Pasteur, M. L. (1858): C. R. Hebd. Seances Acad. Sci. Bd. 46, S. 615-618.
- ↑ W. Marckwald, A. McKenzie, Ber. Dtsch. Chem. Ges. 1899, 32, S. 2130.
- ↑ Emanuel Gil-Av and the Separation of Enantiomers on Chiral Stationary Phases by Chromatography.
- ↑ Reflections on Process Research II, von Edward J. J. Grabowski.
- ↑ Degussa Corporate Citizenship Report 2002.
- ↑ Axel Kleemann und Jürgen Martens: Optical Resolution of Racemic S-(Carboxymethyl)cysteine, Liebigs Annalen der Chemie 1982, 1995−1998.
- ↑ Kurt Günther, Jürgen Martens und Maren Schickedanz: Dünnschichtchromatographische Enantiomerentrennung mittels Ligandenaustausch, Angewandte Chemie 96 (1984) 514-515; Angewandte Chemie International Edition in English 23 (1984) 506.
- ↑ Homepage von Prof. Dr. Volker Schurig.
- ↑ Prof. Dr. Wilfried A. König: Cyclodextrin Chemistry and Enantioselective Gas Chromatography.
- ↑ Bernd Schäfer: Naturstoffe der chemischen Industrie, Elsevier GmbH, Spektrum Verlag, 2007, Seite 155, ISBN 978-3-8274-1614-8.
- ↑ Kinetic resolution of a diltiazem intermediate by lipase-catalyzed enantioselective alcoholysis von Michal Shapira-Levinger, Ayelet Fishman in: Journal of Molecular Catalysis B: Enzymatic 9 2000. 251–257.
- ↑ Stereoselective hydrogenation via dynamic kinetic resolution R. Noyori, T. Ikeda, T. Ohkuma, M. Widhalm, M. Kitamura, H. Takaya, S. Akutagawa, N. Sayo, T. Saito, and et al. J. Am. Chem. Soc.; 1989; 111, 9134 - 9135; doi:10.1021/ja00207a038.
- ↑ Palladium-Catalyzed Dynamic Kinetic Asymmetric Allylic Alkylation with the DPPBA Ligands Barry M. Trost and Daniel R. Fandrick in: Aldrichimica Acta 40, 3 , 2007 [1].
Weblinks
- Resolution of chiral drugs, von William H. Porter in: Pure & Appl. Chem., Vol. 63, No. 8, pp. 11 19-1 122,1991.
- Chiral Separations, von Timothy J. Ward in: Anal. Chem. 2006,78,3947.





Wikimedia Foundation.