- LQTS
-
Dieser Artikel erläutert das Long-QT-Syndrom; es existiert aber auch das (wesentlich seltenere) Short-QT-Syndrom. - Für Männer:
- Für Frauen:
- Haverkamp W et al.: QT-Syndrome: Aspekte zur Pathogenese, molekularen Genetik, Diagnostik und Therapie. Dt Ärztebl (1997) 94:A667-A672
- Haverkamp W et al.: Medikamentenbedingte QT-Verlängerung und Torsade de pointes: Ein multidisziplinäres Problem. Dt Ärztebl (2002)99:A1972-1979
- Crotti L, Celano G, Dagradi F, Schwartz PJ: Congenital long QT syndrome. Orphanet J Rare Dis. 2008 Jul 7;3:18. Review. PMID 18606002
- ↑ Delisle BP et al.: Biology of Cardiac Arrhythmias: Ion Channel Protein Trafficking. Circ Res (2004) 94:1418-1428 PMID 15192037.
- ↑ Koops S: Sterben Sportler plötzlich, ist meist das Herz aus dem Takt geraten. Ärztezeitung 2. Juli 2003, online abgerufen am 21. Januar 06.
- ↑ Löllgen H et al.: Plötzlicher Herztod im Sport. Notfallmed (2003) 29: 148–158.
- ↑ Wedekind H et al.: Sudden infant death syndrome and long QT syndrome: an epidemiological and genetic study. Int J Legal Med (2005) 1-9. PMID 16012827.
- ↑ Priori SG et al.: Risk Stratification in the Long-QT Syndrome. New Engl J Med (2003) 348:1866-1874. PMID 12944579.
- ↑ Westenskow P et al.: Compound Mutations - A Common Cause of Severe Long-QT Syndrome. Circulation (2004) 109:1834-1841. PMID 15051636.
- ↑ Bazett JC. An analysis of time relations of electrocardiograms. Heart 1920; 7:353-67
- ↑ Fridericia LS. Die Systolendauer im Elektrokardiogramm bei normalen Menschen und bei Herzkranken. Acta Med Scand 1920; 53:469-86
- ↑ Azie NE et al.: Comparing methods of measurement for detecting drug-induced changes in the QT interval: implications for thoroughly conducted ECG studies. Ann Noninvasive Electrocardiol (2004) 9(2):166-74. PMID 15084215.
- ↑ Maron BJ et al.: Recommendations for Physical Activity and Recreational Sports Participation for Young Patients With Genetic Cardiovascular Diseases. Circulation (2004) 109:2807-2816. PMID 15184297.
- Wie Stress das Herz aus dem Takt bringt; Auswirkung von Cortisol auf mutierte IKs-Kanäle bei LQT-Syndrom; Scinexx, Springer-Verlag, Heidelberg, 2008
LQTS — (medicine) abbrev Long QT syndrome … Useful english dictionary
LQTS — long QT syndrome * * * LQTS abbr long QT syndrome … Medical dictionary
lqts — laughing quietly to self … Glossary of chat acronyms & text shorthand
LQTS — • long QT syndrome … Dictionary of medical acronyms & abbreviations
LQTS — long QT syndrome …
Long QT syndrome (LQTS) — A genetic (inherited) condition that predisposes individuals to irregular heartbeats (arrhythmias), fainting spells and sudden death. The irregular heartbeats are typically brought on by stress or vigorous activity. LQTS is often symptomless and… … Medical dictionary
QT syndrome, long (LQTS) — A genetic (inherited) condition that predisposes individuals to irregular heartbeats (arrhythmias), fainting spells and sudden death. The irregular heartbeats are typically brought on by stress or vigorous activity. LQTS is often symptomless and… … Medical dictionary
Syndrome, long QT (LQTS) — A genetic (inherited) condition that predisposes individuals to irregular heartbeats (arrhythmias), fainting spells and sudden death. The irregular heartbeats are typically brought on by stress or vigorous activity. LQTS is often symptomless and… … Medical dictionary
Long QT syndrome — Infobox Disease Name = Long QT syndrome(Romano Ward syndrome) Caption = Schematic representation of normal ECG trace (sinus rhythm), with waves, segments, and intervals labeled. DiseasesDB = 11104 ICD10 = ICD10|I|45|8|i|30 ICD9 = ICD9|426.82 ICDO … Wikipedia
ARCM — Klassifikation nach ICD 10 I42. Kardiomyopathie … Deutsch Wikipedia
- Kontaktieren Sie uns: Unterstützung, Werbung
| Klassifikation nach ICD-10 | ||
|---|---|---|
| I45.8 | Sonstige näher bezeichnete kardiale Erregungsleitungsstörungen | |
| ICD-10 online (WHO-Version 2006) | ||
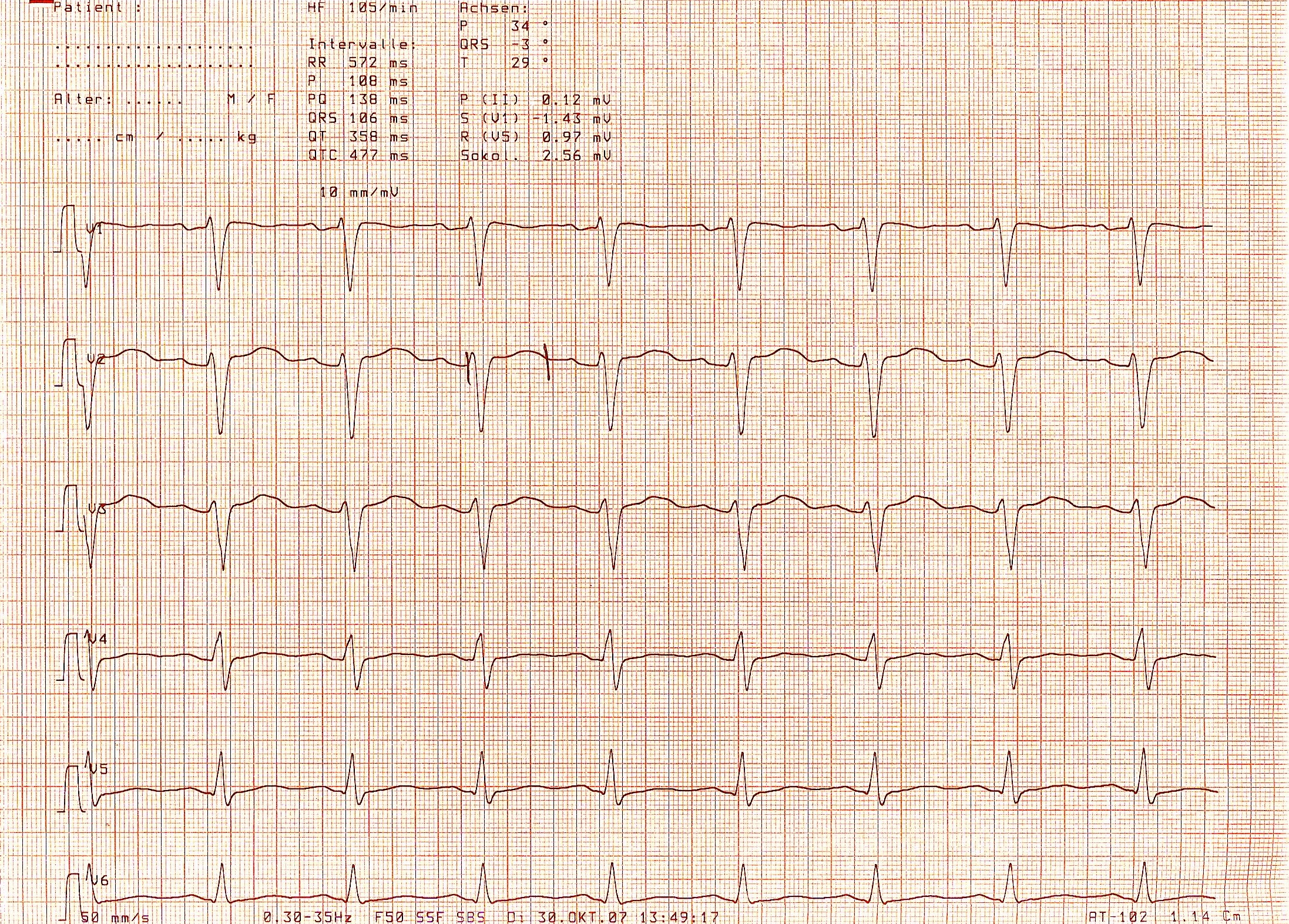
Das Long-QT-Syndrom ist eine seltene, lebensgefährliche Krankheit, die bei sonst völlig herzgesunden Menschen zum plötzlichen Herztod führen kann. Es ist entweder vererbt (kongenital) oder erworben, dann meist als Folge einer unerwünschten Arzneimittelwirkung. Die bekanntesten kongenitalen Long-QT-Syndrome sind das Romano-Ward-Syndrom (syn.: Pseudohypokaliämie-Syndrom) und das Jervell- und Lange-Nielsen-Syndrom (JLNS).
Das wegweisende und namensgebende Krankheitszeichen des Long-QT-Syndroms ist eine Verlängerung der QT-Zeit im Elektrokardiogramm (EKG) mit einer frequenzkorrigierten QT-Zeit (QTc) von über 440 Millisekunden (ms). Für das Long-QT-Syndrom typisch ist anfallsweise auftretendes Herzrasen (Tachykardie), oft in Form der lebensbedrohlichen Torsade de pointes-Tachykardien. Diese Herzrhythmusstörungen können zu Schwindelattacken, plötzlicher Bewusstlosigkeit (Synkope) und zum Herzstillstand durch Kammerflimmern führen. Viele Patienten leiden aber unter keinerlei Beschwerden, bleiben also asymptomatisch.
Sowohl die Tachykardien als auch die Synkopen treten bevorzugt bei körperlicher Belastung oder in Stresssituationen auf. Bei symptomatischen Patienten ist die Prognose ohne Behandlung schlecht, fast allen Patienten kann aber heutzutage eine adäquate Therapie angeboten werden.
Inhaltsverzeichnis |
Ursache und Formen
Pathogenese
Ursache des Long-QT-Syndroms sind geringfügige Abweichungen im Ablauf der elektrischen Signalübermittlung in den Zellen des Herzmuskels (Myokard). Dabei handelt es sich um eine verzögerte Repolarisation, hauptsächlich eine Verlängerung der als Phase 2 bezeichneten Plateauphase des Aktionspotentials. Während dieser früher oft als vulnerable Phase bezeichneten Zeit von etwa 300-400 Millisekunden können irreguläre Nachdepolarisationen bereits wieder ein Aktionspotenzial auslösen, welches dann länger anhaltende Arrhythmien triggern kann („getriggerte Aktivität“). Bei den kongenitalen Long-QT-Syndromen wird die Verlängerung der Plateauphase durch abnorme Eigenschaften der Ionenkanäle verursacht, entweder in Form eines verminderten Ionentransports („loss of function“ des Kalium-Ionenkanals beim LQTS1 und LQTS2) oder einer erhöhten Transportleistung („gain of function“ des Natrium-Ionenkanals beim LQTS3)[1]. Beim erworbenen Long-QT-Syndrom wird sie in erster Linie auf eine Hemmung des schnellen Anteils des Kalium-Ionenstromes IKr zurückgeführt.
Kongenitale Long-QT-Syndrome
Seit Mitte der 60er Jahre des 20. Jahrhunderts wurden mit dem Romano-Ward- und dem Jervell- und Lange-Nielsen-Syndrom zunächst zwei klinische Erscheinungsformen (Phänotypen) des angeborenen Long-QT-Syndroms unterschieden. Heute sind molekularbiologisch eine Vielzahl verschiedener Syndrome identifiziert, wobei aktuell sechs Genotypen (LQTS1-LQTS6) dem Romano-Ward-Syndrom zugerechnet werden, zwei dem JLNS (JLNS1-JLNS2) und einer dem Andersen-Syndrom (LQTS7). Ihnen gemeinsam ist eine Mutation von Genen, welche die Ionenkanäle der Herzmuskelzellen kodieren.
Bei den kongenitalen Formen sollten alle blutsverwandten Familienmitglieder auf das Vorliegen eines QT-Syndroms untersucht werden.
| Syndrom | Genort | Gen | Vererbung | Häufigkeit |
|---|---|---|---|---|
| LQTS1 | 11p15.5 | KvLQT1 (KCNQ1) | dominant | 40-55% |
| LQTS2 | 7q35-36 | HERG (KCNH2) | dominant | 35-45% |
| LQTS3 | 3p21-24 | SCN5a (hNaV1.5) | dominant | |
| LQTS4 | 4q25-27 | ANKB | dominant | sehr selten |
| LQTS5 | 21q22.1–22.2 | MinK (KCNE1) | dominant | |
| LQTS6 | 21q22.1–22.2 | MiRP1 (KCNE2) | dominant | |
| LQTS7 | 21q22.1–22.2 | Kir2.1 (KCNJ2) | dominant | |
| Sporadisches QTS | ? | HERG ? | ? | |
| JLN1 | 11p15.5 | KvLQT1 (KCNQ1) | rezessiv | ca. 6.3% |
| JLN2 | 21q22.1–22.2 | MinK (KCNE1) | rezessiv | ca. 0.7% |
Jervell- und Lange-Nielsen-Syndrom
1957 beschrieben A. Jervell und F. Lange-Nielsen eine achtköpfige Familie in Norwegen, in der vier Kinder taubstumm waren. Mitglieder der Familie fielen wegen wiederholter Schwindelattacken und Bewusstlosigkeiten auf und zeigten im EKG eine deutliche Verlängerung des QT-Intervalls. Drei dieser Kinder verstarben an einem plötzlichen Herztod. Als Ursache wurde später ein autosomal-rezessiv vererbtes (vgl. Vererbung) Syndrom mit Innenohrschwerhörigkeit und QT-Verlängerung identifiziert, das heute als Jervell- und Lange-Nielsen-Syndrom (JLNS) bezeichnet wird. Etwa sieben Prozent der kongenitalen Long-QT-Syndrome werden dem JLNS zugerechnet, circa 0,25 Prozent aller schwerhörigen Kinder leiden am JLNS.
Bei klinisch gesunden Eltern, die aber beide das mutierte Gen tragen, werden statistisch gesehen 25 Prozent der Kinder erkranken, 50 Prozent das Gen ohne eigene Erkrankung tragen und 25 Prozent von dieser genetischen Veränderung frei sein.
Romano-Ward-Syndrom
Bei etwa 70 Prozent der vererbten Long-QT-Syndrome liegt eine der autosomal-dominanten Varianten ohne Hörstörung vor, die nach den erstbeschreibenden Kinderärzten C. Romano und O. Connor Ward als Romano-Ward-Syndrom oder auch als Pseudohypokaliämie-Syndrom bezeichnet werden. Dabei handelt es sich tatsächlich um mindestens sechs molekulargenetisch unterscheidbare Genmutationen, die unterschiedliche Ionenkanäle betreffen und den betroffenen Patienten mehr oder weniger stark gefährden. Bei einem erkrankten Elternteil mit den Zeichen des Syndroms können statistisch gesehen die Hälfte der Kinder erkranken, während die andere Hälfte das Gen nicht trägt. Männliche und weibliche Familienmitglieder sind mit gleicher Häufigkeit betroffen.
Erworbenes Long-QT-Syndrom
Eine Verlängerung des QT-Intervalls im EKG kann auch durch den Einfluss einer Vielzahl von Arzneimitteln, durch Elektrolytstörungen und möglicherweise als Folge von Entzündungen (Myokarditis) und Durchblutungsstörungen (Ischämie) entstehen. Wenn dabei Torsades-de-pointes-Tachykardien oder gar Synkopen auftreten, spricht man von einem erworbenen Long-QT-Syndrom, wobei bis heute unklar ist, inwieweit diese Patienten in Wirklichkeit ein verborgenes, kongenitales Long-QT-Syndrom aufweisen.
In den 60er-Jahren des 20. Jahrhunderts wurden erste Berichte über eine Verlängerung der QT-Zeit durch das damals zur Behandlung von Rhythmusstörungen sehr gebräuchliche Chinidin veröffentlicht. Seit den 90er-Jahren hat das durch Medikamente hervorgerufene Long-QT-Syndrom zunehmende Beachtung gefunden, nachdem immer mehr in dieser Hinsicht gefährliche Substanzen identifiziert wurden. Mittlerweile umfasst diese Liste mehr als hundert zum Teil häufig eingesetzte Präparate der unterschiedlichsten Gruppen, wobei oft nur ein oder zwei Vertreter einer Stoffklasse betroffen sind. Darunter finden sich neben Antiarrhythmika wie Chinidin und Sotalol auch viele häufig verschriebene Medikamente, deren kardiale Nebenwirkung lange Zeit überhaupt nicht bekannt war. In den Fokus geraten sind auch Antibiotika wie Erythromycin und Trimethoprim-Sulfamethoxazol, einige Antihistaminika, viele Neuroleptika, andere Psychopharmaka, Parkinson- und Anti-Malaria-Mittel sowie Röntgenkontrastmittel. Mehrere Präparate (u. a. Grepafloxacin, Astemizol, Droperidol, Cisaprid und Clobutinol) sind deswegen bereits vom Markt genommen worden. Eine ausführliche und aktuelle englischsprachige Liste der beachtenswerten Medikamente wird an der University of Arizona gepflegt (vgl. Weblinks).
Gemeinsam ist diesen Substanzen, dass sie in der Herzmuskelzelle den Kaliumausstrom während der Repolarisation hemmen und so das QT-Intervall verlängern können. Das Risiko für derartige unerwünschte Arzneimittelwirkungen (UAW) ist bei niedrigen Pulsfrequenzen (Bradykardie), weiblichem Geschlecht, erniedrigtem Kaliumspiegel im Blut (Hypokaliämie), Verdickung des Herzmuskels durch arterielle Hypertonie (Bluthochdruck), Herzmuskelschwäche und hohen Wirkstoffkonzentrationen auf Grund pharmakogenetischer Besonderheiten erhöht.
Häufigkeit und Prognose
Plötzliche Todesfälle junger und sonst gesunder Menschen erregen Aufmerksamkeit, besonders wenn sie sich bei großen Sportveranstaltungen vor den Augen der Öffentlichkeit ereignen. In diesem Zusammenhang wird neben Herzinfarkt, Herzmuskelerkrankung und Herzmuskelentzündung oft auch auf das QT-Syndrom als mögliche Ursache hingewiesen[2].
Statistisch gesehen sind derartige Ereignisse aber selten, und noch seltener sind sie Folge eines QT-Syndroms. Insgesamt wird für den plötzlichen Herztod von einer Prävalenz von 1-2 pro 1000 Einwohner und Jahr ausgegangen, bei unter 30-jährigen nur etwa 0,5-1 pro 100.000 Einwohner und Jahr[3]. Etwa sechs Prozent der plötzlich Verstorbenen weisen bei einer Obduktion keine Anzeichen einer organischen Herzkrankheit auf, sind also einer primären Rhythmusstörung erlegen. Es wird angenommen, dass davon etwa ein Drittel ein QT-Syndrom aufwies. Diese zum Teil geschätzten Zahlen lassen vermuten, dass in Deutschland jährlich etwa zehn bis zwanzig Menschen im Alter von unter 30 Jahren an einem QT-Syndrom sterben.
Der Verdacht, auch ein Teil der Fälle von plötzlichem Kindstod könnte durch ein kongenitales QT-Syndrom verursacht sein, konnte in einer Untersuchung von 41 Fällen zumindest molekulargenetisch und anhand der Untersuchung von Familienmitgliedern nicht erhärtet werden[4].
Angeborene QT-Syndrome treten mit einer Häufigkeit von 1:5.000 bis 1:15.000 aller Lebendgeburten auf. Etwa 30-46 Prozent dieser Patienten erleiden vor dem 40. Lebensjahr eine Synkope, unbehandelt versterben etwa 20 Prozent der Patienten im ersten Jahr und 50 Prozent in den ersten fünf Jahren nach dem Auftreten von Symptomen.
Mit Hilfe des Ruhe-EKG und einer molekulargenetischen Untersuchung können heute die Patienten mit einem besonders hohen Risiko besser identifiziert werden. Als Hochrisikogruppe gelten alle Patienten mit einem QTc-Intervall von mehr als 500 ms und den Genotypen LQTS1 und LQTS2 sowie LQTS3 bei männlichem Geschlecht. Sie haben unbehandelt ein Risiko von mehr als 50 Prozent, vor ihrem 40. Lebensjahr eine Synkope, einen Herzstillstand oder den plötzlichen Herztod zu erleiden[5]. Ebenfalls besonders gefährdet sind Menschen, die mehr als eine der bekannten Mutationen aufweisen; damit ist bei knapp acht Prozent der angeborenen QT-Syndrome zu rechnen[6]. Diese als compound mutation bezeichneten Genotypen sind häufiger symptomatisch (100 vs. 72 Prozent) und erleiden häufiger einen Herzstillstand (56 vs. 27 Prozent) als solche mit weniger als zwei nachgewiesenen Mutationen.
Krankheitszeichen
Die Verlängerung der QT-Zeit selbst ist normalerweise nicht spürbar, mehr als die Hälfte der Patienten mit einem Long-QT-Syndrom leiden an keinerlei Beschwerden. Wenn Symptome auftreten, so sind sie bereits durch potentiell lebensbedrohliche (maligne) Herzrhythmusstörungen verursacht, die schon als ein schwerwiegendes Krankheitszeichen gewertet werden müssen. Dabei handelt es sich um anhaltende (> 30 Sekunden) oder nicht-anhaltende (≤ 30 Sekunden) ventrikuläre Tachykardien meist vom Typ der Torsade de pointes-Tachykardie. Je nach Dauer und Pulsfrequenz der Tachykardie, Körperposition und allgemeiner Verfassung können diese Tachykardien gar nicht bemerkt werden, zu Schwindel oder plötzlicher Bewusstlosigkeit (Synkope) oder gar zum Herzstillstand und damit zum plötzlichen Herztod führen.
Da die Tachykardien urplötzlich und bevorzugt bei körperlicher Belastung oder in Stresssituationen auftreten, werden auch die Symptome häufig unerwartet und aus völligem Wohlbefinden in den beschriebenen Situationen bemerkt.
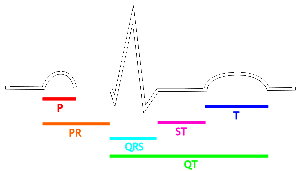
das QT-Intervall ist grün gekennzeichnet
Diagnostik
Der wegweisende und namensgebende Befund des Long-QT-Syndroms ist die Verlängerung des QT-Intervalls im Ruhe-EKG.
Die in Millisekunden (ms) gemessene QT-Zeit ist für sich genommen wenig aussagekräftig, da sie beim Menschen u. a. von der Herzfrequenz, dem Alter und dem Geschlecht abhängig ist. Um eine abnormal lange QT-Zeit zuverlässig erkennen und verschiedene QT-Zeiten im Verlauf miteinander sinnvoll vergleichen zu können, ist eine rechnerische Korrektur der gemessenen QT-Zeit erforderlich. Am häufigsten wird die Bazett-Formel[7] genutzt:
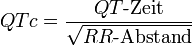 ,
,
wobei die QT-Dauer in ms und der RR-Abstand in Sekunden anzugeben ist. Bei Herzfrequenzen über 100 pro Minute führt die Korrekturformel nach Bazett zu einer Überkorrektur, bei Herzfrequenzen unter 60 pro Minute zu einer Unterkorrektur. Bei Frequenzen über 80 pro Minute führt die in den letzten Jahren zunehmend häufiger angewandte Formel nach Fridericia[8] zu exakteren Ergebnissen:
![QTc={QT\textrm{-Zeit} \over \sqrt[3]{RR\textrm{-Abstand}}}](/pictures/dewiki/99/c48a4f7b9f512b0a72e49b9232bb7ba9.png) ,
,
wobei ebenfalls die QT-Dauer in ms und der RR-Abstand in Sekunden anzugeben ist.
Für wissenschaftliche Zwecke ist eine genauere Korrektur der QT-Zeit erforderlich, die auch das Geschlecht und das Alter des Patienten berücksichtigt. Dies geschieht nach folgenden Formeln:
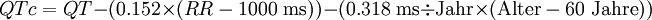
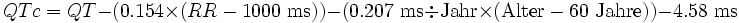
Nachteil der genannten Korrekturformeln ist der erforderliche Rechenschritt, der einen Rechner oder entsprechende Nomogramme erforderlich macht. Aus diesem Grund verwenden viele Ärzte spezielle „EKG-Lineale“, die zur ermittelten Herzfrequenz jeweils die normale QT-Zeit angeben.
Als oberer Grenzwert gilt eine QTc von 440 ms, ab 500 ms ist von einem hohen Risiko auszugehen. Für die Bewertung der „gemessenen“ QT-Zeit und damit auch der berechneten QTc sind die Fehlerquellen der Methode nicht unerheblich. Besonders bei niedrigen Amplituden der T-Wellen und gelegentlich nachfolgenden U-Wellen ist das Ende der T-Welle und damit der Endpunkt der Messung nicht exakt definiert und unterliegt der subjektiven Wahrnehmung des Untersuchers. Darüber hinaus unterscheiden sich die aus einer, drei oder zwölf gleichzeitig abgeleiteten EKG-Linien ermittelten QT-Intervalle signifikant[9], so dass die Messmethode bei Vergleichen berücksichtigt werden sollte.
Abschätzung der QT-Zeit aus dem Ruhe-EKG
Eine verlängerte QT-Zeit kann relativ einfach im Ruhe-EKG erkannt werden, wenn man den RR-Abstand zweier benachbarter QRS-Zacken betrachtet. Ist die QT-Zeit länger als der halbe RR-Abstand, dann ist die QT-Zeit auf jeden Fall verlängert.
Therapie
Da die Häufigkeit schwerwiegender Herzrhythmusstörungen unter einer Behandlung mit Betarezeptorenblockern eindeutig abnimmt, gehören sie zur Standardtherapie bei kongenitalem Long-QT-Syndrom. Patienten, bei denen trotzdem noch Synkopen auftreten und solche nach einem überlebten Herzstillstand sollten vorsorglich einen implantierbaren Defibrillator (ICD) erhalten. Möglicherweise profitieren Patienten mit einem besonders hohen Risiko bereits vor dem Auftreten von Symptomen von der Implantation eines ICD.
Bei einem durch Medikamente verursachten Long-QT-Syndrom steht das unverzügliche Absetzen der Substanz im Vordergrund. Betablocker gelten - im Gegensatz zur kongenitalen Form - hier als kontraindiziert, da sie eine Bradykardie hervorrufen oder verstärken und so das Risiko bedrohlicher Rhythmusstörungen eher erhöhen. Bewährt hat sich neben dem Ausgleich einer evtl. Hypokaliämie die Zufuhr von Magnesium, bei Bradykardie wird eine Steigerung der Herzfrequenz durch Medikamente (z. B. Orciprenalin) oder eine vorübergehende Schrittmacherstimulation empfohlen.
Körperliche Belastung ist für Patienten mit einem Long-QT-Syndrom nicht unproblematisch. Besonders bei abruptem Belastungsbeginn oder -ende, Kälte, Druckschwankungen und lauten Geräuschen besteht ein erhöhtes Risiko für bedrohliche Rhythmusstörungen. Aus diesem Grund wird von Sportarten wie Basketball, Eishockey, Bodybuilding, Surfen, Schwimmen, Tauchen und Schnorcheln grundsätzlich abgeraten, ebenso vom wettkampfmäßigen Laufen, Gewichtheben, Motorradfahren, Squash- und Tennisspielen. Regelmäßige, moderate, körperliche Aktivität wie Joggen, Walking und Skaten hingegen wird befürwortet, und auch gegen Bowlen, Tennisspielen und Gewichtheben ist wenig einzuwenden, wenn es nicht leistungsorientiert erfolgt[10]. Kinder werden oft vom Schulsport befreit, da dieser unter dem Aspekt der Benotung (Gefahr einer Überforderungssituation) steht, individuell kann aber über die Ausübung von anderen Freizeitsportarten entschieden werden.
Literatur
Quellen
Weblinks
|
Wikimedia Foundation. Schlagen Sie auch in anderen Wörterbüchern nach:18+
© Academic, 2000-2026
Wörterbücher Export, schritte mit PHP, Joomla, Drupal, WordPress, MODx.
|