- Chemismus der Dipeptidylpeptidase 4
-
Das Enzym Dipeptidylpeptidase 4 (kurz DPP 4, DP IV oder DPP IV) wurde erstmals 1964/66 von Hopsu-Havu beschrieben. Erst im Laufe der Zeit wurde erkannt, dass dieses Enzym wesentliche Funktionen im menschlichen und tierischen Organismus ausübt.[1]
Nach der Nahrungsaufnahme werden im Magen verschiedene Hormone freigesetzt, sogenannte Inkretine, die die Freisetzung von Insulin im Körper steigern. Die DPP 4 spaltet diese Inkretine und stoppt somit deren Wirkung. Daraus folgt, dass Inhibitoren der DPP 4 potentielle Medikamente gegen Diabetes mellitus Typ II sind, da eine vermehrte Freisetzung von Insulin helfen kann, den chronisch erhöhten Blutzucker zu senken. Als Ergebnis der Erforschung der Dipeptidylpeptidase-4 und deren Inhibitoren sind bereits zwei Arzneistoffe, Sitagliptin und Vildagliptin, durch den Ausschuss für Humanarzneimittel (CHMP) der Europäischen Arzneimittelagentur als Therapeutika zur Behandlung des Diabetes mellitus Typ 2 zugelassen worden.[2]
Gegen Diabetes mellitus Typ I würde eine Hemmung dieses Enzyms nichts bewirken: Hier kann überhaupt kein Insulin mehr produziert werden, da die Inselzellen der Bauchspeicheldrüse vollkommen zerstört sind.
Der Funktionsmechanismus aus chemischer Sicht (Chemismus)
Das Enzym DPP 4 kommt ubiquitär im menschlichen, aber auch im tierischen Organismus vor. Es ist eine Proteinase, die vom N-terminalen Ende eines Peptids Zweiergruppen von Aminosäuren (Dipeptide) abspaltet. Da sich im Aktiven Zentrum des Enzyms die Aminosäure Serin befindet, gehört es zu den Serinproteinasen. Es kann nur vom Aminogruppen-Ende (N-Terminus) eines Peptids Dipeptide abspalten, nicht jedoch vom Carboxygruppen-Ende, dem C-Terminus (siehe Peptidbindung).
Ein beispielhaftes Peptid als Substrat für die DPP 4 sieht folgendermaßen aus, wobei Xaa2, Xaa1 und X'aa1 für beliebige Aminosäuren stehen und der senkrechte Strich für die Stelle, an der das Enzym die Peptidbindung spaltet. Mit P2, P1 und P'1 werden die Positionen der Aminosäuren relativ zur Spaltungsstelle beschrieben:
N-Xaa2-Xaa1-|-X'aa1-… P2 P1 P'1 N-Xaa2-Xaa1-C + N-X'aa1-…
Die DPP 4 spaltet bevorzugt Peptide, wenn sich ein Prolinrest (Pro) in P1-Position befindet.[3]
Die Substratspezifität in P1-Position (Xaa1)
Neben Peptiden mit Prolin in P1-Stellung, spaltet die DPP 4 auch noch Peptide mit anderen Aminosäuren in dieser Position.[4][5]
Zunächst wurde festgestellt, dass neben Prolin- auch Hydroxyprolin- und Dehydroprolinreste sehr gut akzeptiert werden. Auch Substrate mit Pipecolinsäureresten (Pip) in P1-Stellung sind ähnlich aktiv. Im Schema 1 wird ein „Prolinstammbaum“ vorgestellt [6] der auf direktem Wege vom Prolin bis zum Glycin führt. Versuchsweise wurden die einzelnen Di-Peptid-para-Nitroanilide (pNA) in P1-Stellung gegen die angeführten Aminoacylreste des „Stammbaums“ ausgetauscht. Hierbei konnte gezeigt werden, dass alle diese Derivate mehr oder weniger aktive Substrate darstellen. Erfolgreich geprüft wurden in Ala-Xaa1-pNA besonders Xaa1 = Thz, Oxa, Pipecolinsäure (Pip) und Azetidin-2-carbonsäure (Aze).
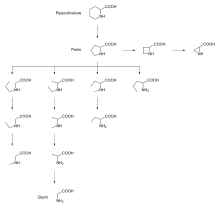 Schema 1: Prolinstammbaum (In dem Schema ist auch das Sechsringsystem der Pipecolinsäure enthalten, sie liefert auch aktive Substrate).
Schema 1: Prolinstammbaum (In dem Schema ist auch das Sechsringsystem der Pipecolinsäure enthalten, sie liefert auch aktive Substrate).
Ferner wurden geprüft die Xaa1-Reste von: Glycin (Gly), 2-Aminobutansäure (Abu), Norvalin (Nva), Sarcosin (Sar), N-Ethyl-Glycin, N-(n-Propyl)-Glycin, N-Methyl-2-Aminobutansäure, N-Methyl-Alanin und N-Ethyl-Alanin. Interessant ist, dass es im Hinblick auf die Wirksamkeit der DPP 4 Substrate eine Abstufung in folgender Richtung gibt:
- Prolin > Alanin > Glycin.
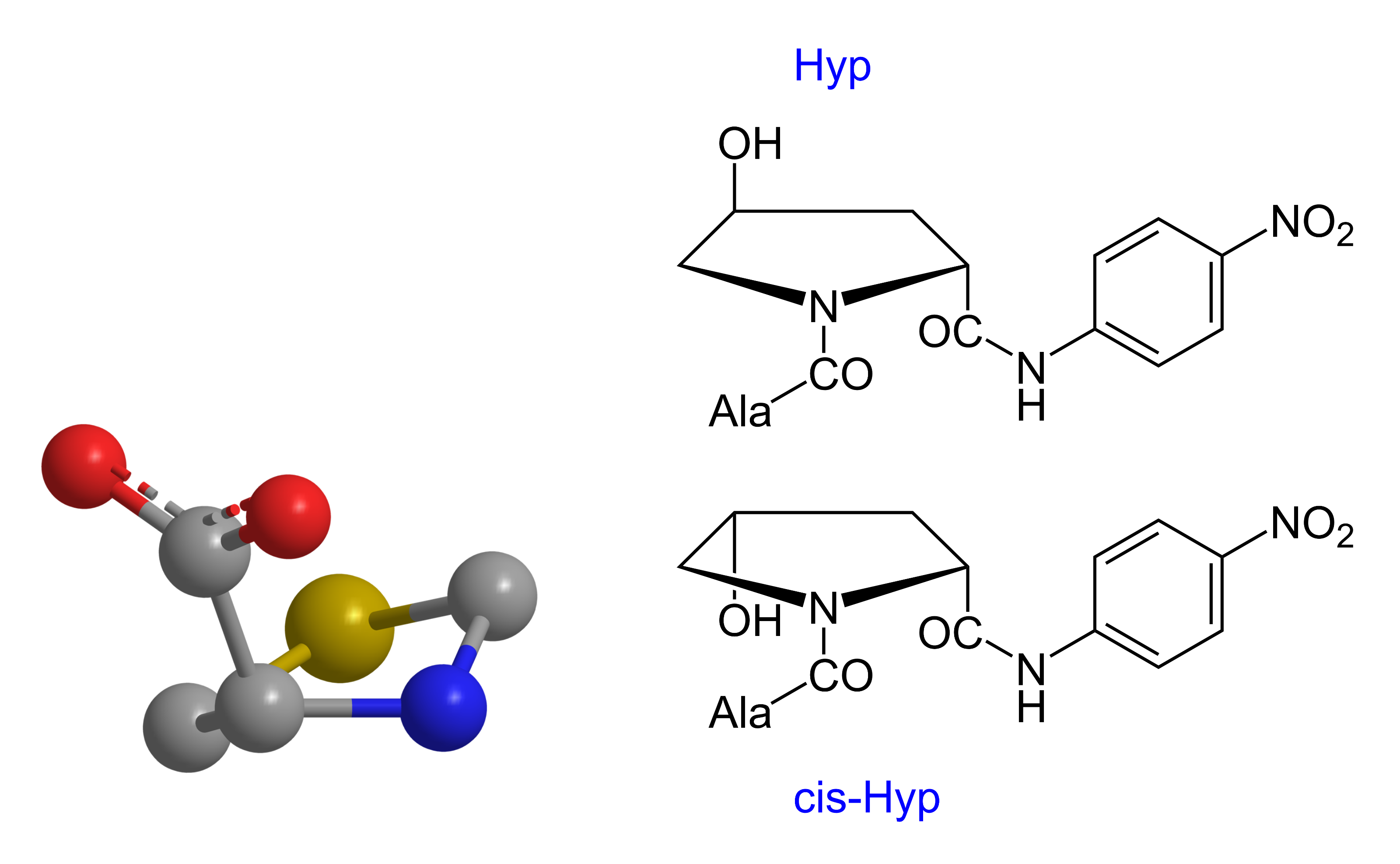
Hinsichtlich des kcat/KM-Wertes ist das Aze-Derivat vergleichbar mit dem entsprechenden Pro-Substrat. In dem einen Fall handelt es sich um einen Fünf- im anderen Fall um einen Vierring. Ebenfalls interessant war die Prüfung der Verbindungen Ala-Xaa1-pNA mit Xaa1 = a-5MeOxa, und s-5MeOxa. Ala-(s-5MeOxa)-pNA fungierte als Substrat, Ala-(a-5MeOxa)-pNA nicht [s = syn, a = anti]. Der Prolinring ist nicht planar gebaut, sondern liegt in einer Konfiguration analog dem Schema 2 (links) vor.
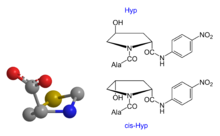 Schema 2: „γ-Puckering“[7] der fünfgliedrigen Ringe am Beispiel des Thz; Oxa = Ersatz von S durch O, Pro = Ersatz von S durch >CH2 (links). Stellung der Hydroxylgruppe im Hyp-System {Ala-Hyp-pNA [Ala-trans-Hyp-pNA oder Ala-anti-Hyp-pNA; Ala-cis-Hyp-pNA oder Ala-syn-Hyp-pNA (rechts)}
Schema 2: „γ-Puckering“[7] der fünfgliedrigen Ringe am Beispiel des Thz; Oxa = Ersatz von S durch O, Pro = Ersatz von S durch >CH2 (links). Stellung der Hydroxylgruppe im Hyp-System {Ala-Hyp-pNA [Ala-trans-Hyp-pNA oder Ala-anti-Hyp-pNA; Ala-cis-Hyp-pNA oder Ala-syn-Hyp-pNA (rechts)}Demzufolge sind auch die Positionen im Ringsystem des Prolins unterschiedlich zu bewerten. Daraus erklärt sich, warum Xaa2-Hyp-pNA ein Substrat ist, Xaa2-(a-5MeOxa)-pNA aber mittels DPP 4 nicht enzymatisch hydrolysiert wird.
Es wird beobachtet, dass in Substraten vom Typ Xaa2-Pro-X’aa1 die Spaltungsrate von der Natur des Restes X’aa1 abhängt. Offensichtlich verschiebt sich in diesem Falle der geschwindigkeitsbestimmende Schritt (siehe unten) von der Deacylierung (k3) in vorangehende Schritte. Denkbar ist eine Behinderung des Einstroms dieser Substrate in das aktive Zentrum der DPP 4. Der langsamste Schritt sollte hier möglicherweise vor dem eigentlichen katalytischen Prozess liegen. Hier ist im einfachsten Fall (bei Annahme von ka sehr viel kleiner als k3 und k3 sehr viel kleiner als k2 und k1)[8]
- kcat = k3ka und M = k-ak3/kak1
ka = Geschwindigkeitskonstante des Einstroms des Substrates in das aktive Zentrum, k-a = Geschwindigkeitskonstante des Ausstroms des Substrates aus dem aktiven Zentrum, k1 = Geschwindigkeitskonstante tetraedrischen Intermediats, k2 = Geschwindigkeitskonstante des Acylierungsschrittes (TI1 ® Acylenzym), k3 = Geschwindigkeitskonstante des Deacylierungsschrittes (TI2 ® Dipeptid).
Die Substratspezifität in P2-Position (Xaa2)
Auch wurde die Bedeutung des N-terminalen Aminosäurerestes in P2-Stellung (Xaa2) eruiert. Untersucht wurden u. a. in Xaa2-Pro-pNA die Reste von: Pro, Abu, Leu, Val, Ala, Ile, Glu, Phe, Tyr, Ser, Gln, Lys, Asp, Asn sowie N,N-Dimethyl-Glycin [(N,N)-DMG], N,N,N-Trimethyl-Glycin [(N,N,N)-TMG] und S,S-Dimethyl-sulfonium-essigsäure [(S,S)-DMS]. Alle Substanzen fungierten als Substrate der DPP 4 (siehe Tabelle 1).
Tabelle 1 X-Pro-p-nitroanilid
X =kcat in s−1 Km in 105 M Pro 51,5 ± 0,3 0,92 ± 0,03 α-Aba 72,7 ± 0,7 1,53 ± 0,05 Leu 65,4 ± 0,6 44,9 ± 0,9 Val 1,75 ± 0,04 1,28 ± 0,08 Ala 54,6 ± 0,4 1,66 ± 0,05 Ile 28,5 ± 4,9 39,5 ± 0,3 Glu 1,23 ± 0,67 2,10 ± 0,07 Phe 71,3 ± 1,7 4,27 ± 0,25 Tyr 62,9 ± 2,2 4,03 ± 0,34 Ser 61,0 ± 0,8 3,99 ± 0.24 Gln 69,8 ±4,7 4,90 ± 0.98 Lys 54,8 ± 1,2 5,16 ± 0,25 Gly 78,4 ± 0,7 10,2 ± 0,52 Asp 30,1 ± 0,3 5,85 ± 0,22 Asn 51,4 ± 1,0 11,8 ± 0,83 Sar 97,3 ± 0,02 13,0 ± 0,12 (N,N)-DMG 0,9 ± 0,02 145 ± 10 (N,N,N)-TMG 1,5 ± 0,2 7040 ± 1200 (S,S)-DMS 0,3 ± 0,02 1135 ± 80 Wie weit entfernt kann die N-terminale Aminofunktion von der Peptidbindung zwischen P1 und P2 sein? Dazu wurden in Xaa2-Pro-pNA Aminosäurereste von Ala, β-Ala, γ-Abu und ε-Ahx verwendet. Die Derivate von Ala, β-Ala und γ-Abu sind Substrate der DPP 4 mit fallender Tendenz. Die ε-Ahx-Verbindung wird nicht hydrolysiert[9]
Tabelle 2 X-Pro-p-nitroanilid
X =kcat in s−1 Km · 105 in M Ala 54,6 ± 0,4 1,66 ± 0,05 β-Ala 6,7 ± 0,1 154 ± 8,5 γ-Abu 0,9 ± 0,03 352 ± 30 ε-Ahx keine hydrolytische Aktivität Die Rolle der Aminoacylreste in P’1-Position (X’aa1)
Um die Bedeutung der Aminoacylreste in P’1-Position zu klären, wurden kinetische Studien mit den Tripeptiden Ala-Pro-X’aa1 durchgeführt. Überraschenderweise zeigt sich, dass keine enzymatische Hydrolyse stattfindet, wenn X’aa1 = Pro-, Hyp-, Dehydroprolyl-, Pipecolyl-, Aciridyl-Reste sind oder allgemein die zu spaltende Peptidbindung –CO–NH– durch –CO–N(R)– ersetzt ist (R ungleich H).
Die „Erkennungsstruktur“ der Substrate der DPP 4
Auf Grund des umfangreichen kinetischen Datenmaterials wurden intensive Studien der Computersimulation durchgeführt. Ein Resultat dieser Bemühungen war die Formulierung einer Struktur, die alle Substrate besitzen müssen, um im aktiven Zentrum der DPP 4 als Substrate erkannt zu werden. Diese „Erkennungsstruktur“ ist in der Abbildung 1 gezeigt.
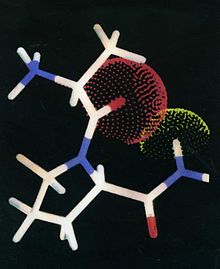 Abb. 1
Abb. 1Von Bedeutung ist die interne H-Brücke. Jede Behinderung ihrer Ausbildung führt zur Inaktivität als Substrat,.[10] Die Tatsache, dass jede Substitution an der NH-Gruppe der zu spaltenden Peptidbindung diese verhindert, ist der Grund, warum alle Peptide die das Strukturmerkmal –CO–N(R)– mit R ungleich H haben einer enzymatischen Hydrolyse nicht zugänglich sind. Es ist zu erwarten dass die interne H-Brücke verhindert wird durch:
- eine cis-Peptidbindung und
- den optischen Antipoden
Es ist bekannt, dass Prolinpeptide über eine messbare cis-Konfiguration an der zu spaltenden Peptidbindung verfügen. Wir haben das Verhältnis von trans- zu cis-Bindung am Beispiel des Substrates Ala-Pro-pNA ermittelt und die enzymatische Hydrolyse mit Hilfe der schnellen Kinetik („Stopped-Flow-Methode“) verfolgt. Es zeigt sich ein biphasischer Umsatz nach der Zeit. In der ersten Phase kann ein schneller von der Enzymkonzentration abhängiger Abbau bis zu dem Prozentsatz, der für das Vorhandensein der trans-Peptidbindung gefunden wurde, beobachtet werden, danach erfolgt ein langsamer Reaktionsverlauf, der bestimmt wird, von der Enzym-unabhängigen cis-trans-Umwandlungsgeschwindigkeit. Demnach erfolgt erwartungsgemäß eine Spaltung nur über die trans-Bindung, die cis-Bindung ist inaktiv.
Die Stereospezifität
Ferner sind die Substrate in P1-Position absolut stereospezifisch. Ala-L-Pro-pNA z. B. ist ein gutes Substrat, Ala-D-Pro-pNA wird enzymatisch nicht hydrolysiert. Bei Verbindungen vom Typ D-Xaa2-Pro-pNA findet ebenfalls keine Hydrolyse statt. D-Xaa2-Ala-pNA aber sind Substrate der DPP 4. Tabelle 3 demonstriert dies am Beispiel des Ala-Ala-pNA, D-Ala-Ala.pNA und Aib-Ala-pNA. Das Phänomen, dass D-Phe-Pro-pNA und D-Tyr-Pro-pNA unkompetitive Inhibitoren der Hydrolyse von Ala-Pro-pNA sind (die Ki-Werte sind: 0,35 ± 0,04 mM und 0,52 ± 0,04 mM), wonach beide Inhibitoren keine kompetitive oder gemischte Hemmer sind, und bei Ala-Ala-pNA als Substrat die Verbindungen keinen inhibitorischen Effekt zeigen, ist im Zusammenhang mit den in dieser Arbeit diskutierten theoretischen Schlussfolgerungen zu sehen.
Tabelle 3: stereospezifische Effekte Verhältnis in KM in kcat in kcat / KM [a] / [c] 0,92 ± 0,13 18,27 ± 1,81 19,95 ± 0,91 [a] / [b] 0,86 ± 0,15 40,39 ± 4,28 47,33 ± 2,47 [c] / [b] 0,95 ± 0,20 2,23 ± 0,34 2,38 ± 0,15 [a] = Ala-Ala-pNitroanalinid, [b] = D-Ala-Ala-pNitroanalinid, [c] = Aib-Ala-pNitroanalinid Auffallend ist, dass die Stereoselektivität in P2, wie die Tabelle 3 zeigt, sich nicht auf den KM-Wert, wohl aber gravierend auf die kcat-Werte aufwirkt. Auf diesen Effekt, der überraschend ist, soll weiter unten eingegangen werden.
Die Sequenz der Enzymkatalyse
Es wird angenommen, dass im Verlaufe der Enzymkatalyse die Reaktionsfolge, dargestellt in Schena 3, durchlaufen wird:
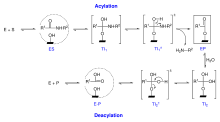 Schema 3
Schema 3Danach ist zwischen einem Acylierungsprozess und einem Deacylierungprozess zu unterscheiden. Das Substrat dringt in das aktive Zentrum ein (siehe Erkennungskomplex) und es bildet sich ES. Daraus folgt das tetraedrische Intermediat des Acylierungsprozesses TI1. Nach Abspaltung des ersten Spaltproduktes entsteht durch Anlagerung eines Wassermoleküls das zweite tetraedrische Intermediat TI2 (Deacylierungsprozess). Nach Spaltung von diesen wird letztendlich das freie Enzym und das zweite Spaltprodukt (Dipeptid) freigesetzt. Das erste tetraedrische Intermediat TI1 ist asymmetrisch. Theoretisch kann sich hier eine S- und/oder eine R-Form ausbilden. TI2 ist dagegen symmetrisch. Hier entstehen keine antipodischen Strukturen.
Zunächst erhebt sich die Frage, wo bei den Substraten der geschwindigkeitsbestimmende Schritt lokalisiert ist, ob im Bereich der Acylierung oder der Deacylierung. Es wurden daher Untersuchungen mit den Substraten Ala-Ala-anilid und Ala-Pro-Anilid mit verschieden substituierten Arylringen im pH-Optimum durchgeführt. Der Hansch-Approach (QSWA = Quantitative-Struktur-Wirkungs-Analyse) brachte keine Korrelaton bei den substituierten Derivaten der Reihe Ala-Pro-Anilid, wohl aber eine solche in der Reihe Ala-Ala-Anilid.
Substrat : Ala-Ala-NH-C6H4-R QSWA : lgkcat = 0,807s ± 0,186 R : -H, p-F, p-Cl, p-Br, p-CH3, p-OCH3, p-OC2H5, p-NO2, m-Cl, m-CH3, m-CF3, m-NO2 Substrat : Ala-Pro-NH-C6H4-R QSWA : keine Korrelation
Daraus geht hervor, dass der geschwindigkeitsbestimmende Schritt bei der Alaninreihe in der Acylierung liegt, in einem Bereich also, wo die substituierten Aniline noch nicht abgespalten sind (k2). In der Prolinreihe dagegen sollte der geschwindigkeitsbestimmende Schritt in der Deacylierung liegen (k3). Die Messungen wurden beim pH-Optimum (siehe unten) durchgeführt.
Die Frage ist nun: lässt sich die Konformation des tetraedrischen Intermediats TI1 ermitteln? Zu diesem Zwecke wurde eine QCAR Analyse (Quantitative Conformation Activity Relationships) in der Alaninreihe durchgeführt. Die Resultate lassen vermuten, dass die Effekte dann auftreten, wenn der –NH-Ar-Rest aus dem tetraedrischen Intermediat TI1 austritt (k2). Aus der QCAR-Analyse folgt nämlich eine mögliche Konformation mit einer „Behinderung“ der Wasserstoffatome a und b, die, entsprechend der Struktur in Schema 4 bei der Rotation des aromatischen Ringes auftritt.
 Schema 4
Schema 4Das Schema 5 demonstriert die beiden Antipoden des TI1. Nur die Form links führt zur sterischen Hinderung der oben genannten Wasserstoffatome, nicht aber die rechts dargestellte Struktur. Dies könnte ein Hinweis auf die Stereospezifität des tetraedrischen Intermediates TI1 sein (im Zusammenhang mit den Ergebnissen der D-Isotopemessungen – siehe unten).
Die Untersuchungen zur pH-Wert Abhängigkeit, demonstriert an der DPP 4 aus Schweinenierenrinde, ergab ein pH-Optimum von ca. 6,7. Das Temperaturoptimum liegt bei etwa 30 °C. Die Studien ergaben ferner, dass für die Aktivität der Substrate eine positive Ladung am N-Terminus nötig ist. Dabei ist die Protonierung der N-terminalen primären Aminogruppe notwendig. Sie ist aber keine Bedingung. Für die Substraterekennung ist die Existenz einer positiven Ladung genügend, wie aus der Tabelle 1 hervorgeht. Streng genommen handelt es sich bei der DPP 4 also nicht um eine Aminopeptidase, sondern um eine Oniumacylaminoacyl-Peptidase. Die katalytische Abspaltung von Dipeptiden aus einem Oligo- oder Polypeptid vom N-terminalen Ende ist aber, physiologisch gesehen, ein essentieller Prozess.
 Schema 5
Schema 5Sekundäre D-Isotopieeffekte und Solventisotopieeffekte in D2O
Im Schema 4 und Schema 5 (links) ist ein mögliches Modell des TI1 dargestellt. Seine Bestätigung sollte durch D-Isotopieeffekte möglich sein, wenn einerseits ein H/D-Austausch im Asymmetriezentrum der P1 Aminosäure erfolgt und andererseits ein H/D-Austausch im aromatischen Ring in o-Stellung erfolgt. Es zeigt sich, dass in beiden Fällen sekundäre D-Isotopieeffekte zu messen sind.
Bei Ala-Ala-pNA (L-Ala-L-Ala-d1-pNA) sind im pH-Optimum bei Zimmertemperatur sekundäre D-Isotopieeffekte messbar. Sie betragen in kcat = 1,27 und in KM = 1,24. Wird das Substrat im aromatischen Ring vollständig deuteriert (Ala-Ala-NH-C6D4-pNO2), so ergeben sich sekundäre D-Isotopieeffekte in kcat und in KM von 1,05 ± 0,03.
Interessant ist, dass ein sekundärer D-Isotopieeffekt in kcat/KM, entsprechend dem Schema 6 in Abhängigkeit vom pH-Wert auftritt. Der Kurvenverlauf ist bemerkenswert. Bei fallenden pH-Werten von etwa 7,5 bis etwa 5,0 ist kein Isotopieeffekt messbar. Ab pH 5,0 beobachtet man einen steigenden sekundären D-Isotopieeffekt mit fallendem pH-Wert, verbunden mit einem Übergang von k3 nach k2 als geschwindigkeitsbestimmenden Schritt.
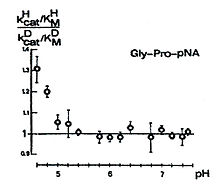 Schema 6
Schema 6Dieses Resultat unterstreicht die hohe Wahrscheinlichkeit der in dem Schema 4 dargestellten Struktur.
Messungen der Solvent-D-Isotopieeffekt ergeben im Falle der Substrate Ala-Pro-pNA und Gly-Pro-pNA mit dem in der Deacylierung liegenden geschwindigkeitbestimmenden Schritt einen Ein-Protonen-Übergang und beim Ala-Ala-pNA, der geschwindigkeitsbestimmende Schritt in der Acylierung liegend, einen Zwei-Protonen-Übergang (Tabelle 4).
Tabelle 4: Zwei-Protonen-Übergang Substrat pH beste Anpassung an die Funktion

(signifikant entsprechend F-Test)Parameter Protonenübergänge Ala-Pro-pNA 7,5 

kcat
kcat/KM1
1Gly-Pro-pNA 6,8 

kcat
kcat/KM1
1Ala-Ala-pNA 7,5 

kcat
kcat/KM(2)
2Inhibitoren
Es wurden eine Reihe von Dipeptiden auf ihre Wirkung als (kompetitive) Inhibitoren der DPP 4 untersucht. Die Tabelle 5 zeigt eine Übersicht.
Tabelle 5: Dipeptide Dipeptide Ki in M Gly-Pro (1,6 ± 0,2) · 10−3 Val-Pro (1,0 ± 0,1) · 10−5 Leu-Pro (7,0 ± 0,7) · 10−5 Ile-Pro (6,9 ± 0,3) · 10−6 Asp-Pro (2,0 ± 1,5) · 10−4 Arg-Pro (4,2 ± 1,0) · 10−5 Phe-Pro (4,7 ± 1,5) · 10−5 ε-Z(4-NO2)Lys-Pro (1,1 ± 0,2) · 10−6 β-Ala-Pro (1,7 ± 0,1) · 10−2 Ala-Ala (4,1 ± 0,7) · 10−4 Ile-Ala (1,0 ± 0,1) · 10−5 Ala-D-Ala (1,8 ± 0,2) · 10−3 Pro-Gly (9,0 ± 0,4) · 10−3 Gly-Phe (3,2 ± 0,5) · 10−3 Ile-Val (4,0 ± 0,1) · 10−5 ε-Z(4-NO2)Lys-D-Pro (4,4 ± 0,3) · 10−5 ε-Z(4-NO2)Lys-Pip (2,2 ± 0,1) · 10−5 ε-Z(4-NO2)Lys-D-Pip (2,0 ± 0,1) · 10−4 ε-N-CapronylLys-Pro (4,8 ± 0,1) · 10−5 ε-N-PalmitylLys-Pro (8,1 ± 0,1) · 10−5
Von diesen Dipeptiden, die auch Spaltprodukte DPP 4-Katalyse sind, ist das Ile-Pro mit einem Ki-Wert von etwa 7,0 · 10−6 M und das ε-Z(4-NO2)Lys-Pro (Ki etwa 10−6) interessant. Um etwa eine Größenordnung wirksamer sind die entsprechenden Pyrrolidide (Tabelle 6).Tabelle 6: Pyrrolidide X-Pyrrolidid
X =pH Ki in M Ile 6,3 (3,3 ± 0,6) · 10−7 Ile 7,6 (1,9 ± 0,4) · 10−7 Phe 6,3 (2,5 ± 0,7) · 10−6 Phe 7,6 (2,3 ± 0,2) · 10−6 ε-Z(4-NO2)Lys 6,3 (3,9 ± 0,5) · 10−7 Röntgenkristallstruktur der Dipeptidylpeptidase 4
Gegenwärtig sind intensive Studien auf den Gebiet der Röntgenkristallstrukturanalyse der DPP 4 im Gange. Das Enzym besteht in der Regel aus zwei identischen Untereinheiten. Jede dieser Untereinheiten besitzt eine N-terminale Peptidsequenz, die das Protein an der Oberfläche einer Zelle verankert. Es wurden auch höhere Aggregate bei der löslichen DPP 4 gefunden, bestehend aus vier identischen Untereinheiten.
Für bestimmte Aussagen ist eine nicht so in das Detail gehende Darstellung der Conolli-Oberfläche günstiger. Hier zeigt sich, dass die DPP 4 eine ungewöhnliche Oberflächenstruktur besitzt. Das aktive Zentrum befindet sich nicht an der Oberfläche außen, sondern in Innern des Proteins. Der Zugang zu dem aktiven Zentrum ist durch einen „Schlauch“ möglich, der eine größere und eine kleinere Öffnung besitzt. Die Frage ist, wie und vor allem wo erfolgt z. B. der Substrateintritt?
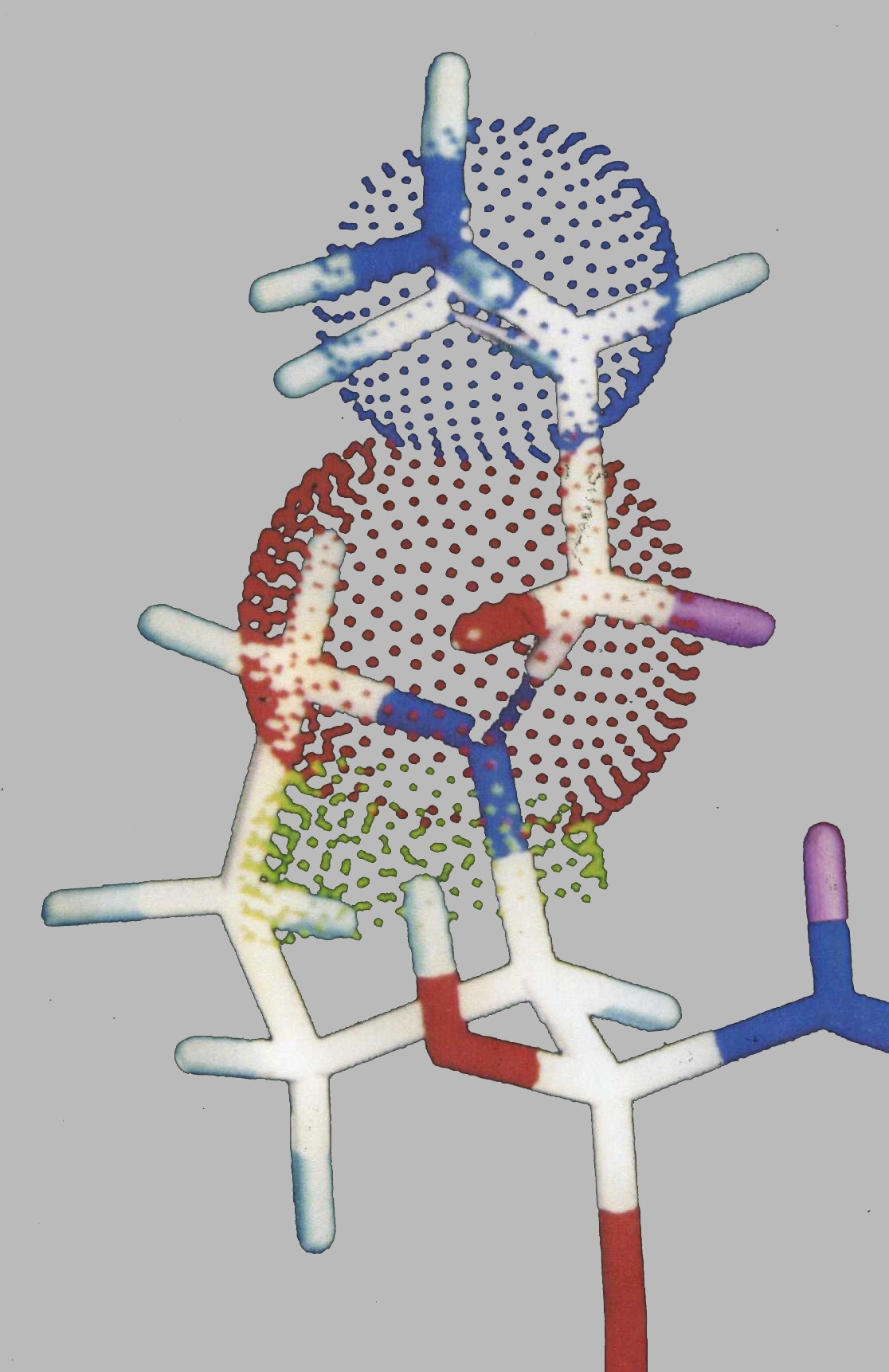
Wenn man sich das elektrostatische Potential an der Oberfläche ansieht, so stellt man fest, dass in der Nähe und innerhalb der großen Öffnung ein sehr starkes negatives Potential mit einem Gradienten in Richtung der Öffnung vorhanden ist (Abbildung 3). Die kleinere Öffnung hat demgegenüber kein so starkes negatives Potential. Es ist daher denkbar, dass die Substrate mit dem positiv geladenen N-Terminus der das Potential größerer Bereiche der Erkennungsstruktur der Substrate prägt in diese Öffnung „hineingezogen“ werden. (Darstellung einer Untereinheit, humane DPP 4).
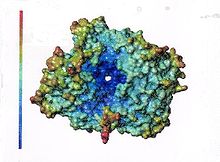 Abb. 3
Abb. 3Die Struktur des tetraedrischen Intermediats TI1
Aus dem experimentellen Befund, wonach das als Substrat fungierende D-Ala-Ala-pNA über kcat wirkt, wobei KM gegenüber dem des Substrates Ala-Ala-pNA unbeeinflusst bleibt (siehe Tabelle 3), drängt sich die Deutung auf, dass der N-terminale Aminoacylrest (wahrscheinlich mit der positiven Ladung) direkt mit dem tetraedrischen Intermediat in P1-Position in Wechselwirkung tritt. Daher wurde eine hypothetische Konformation wie folgt konstruiert (Schema 7).
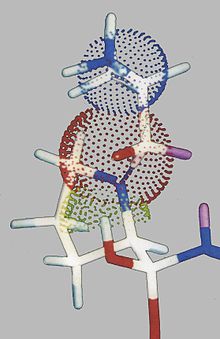 Schema 7
Schema 7
Es scheint zwei Typen von Wasserstoffbrücken zu geben, nämlich solchen, die für die direkte Bindung im aktiven Zentrum verantwortlich sind, d. h., das Substrat oder den Inhibitor an die wechselwirkungsaktiven Reste des Proteins heranführen und solchen, die direkter Bestandteil des Funktionsmechanismus sind. Wenn im Laufe des Funktionsmechanismus der Serinproteasen tetraedrische Intermediate TI1 und TI2 (siehe Schema 3) entstehen, ist TI1 asymmetrisch. Beim Aufrichten des Carbonylatoms der zu spaltenden Peptidbindung können sich zwei antipodische Konformationen ausbilden, von der eine das Peptid als Substrat (1), die andere als Inhibitor (2) erkennen sollte (Schema 5).Die Entscheidung, in welcher Richtung sich die Carbonylgruppe aufrichtet, d. h., welches der antipodischen tetraedrischen Intermediate bevorzugt entsteht, wird davon abhängen, in welcher Richtung die Wechselwirkungen der Proteinseitenketten im aktiven Zentrum (z. B. H-Brücken) am wirkungsvollsten sind bzw. die räumliche Anpassung des Effektormoleküls am effektivsten erfolgt. So ist es durchaus möglich, dass ein Inhibitormolekül auch Substrat sein kann. Eine entscheidende Triebkraft der Abtrennung eines Molekülteils aus dem tetraedrischen Intermediat ist, ob das Prinzip der stereoelektronischen Kontrolle nach Delongchamps[11] gültig ist. Es besagt, dass die Spaltung von Estern oder Amiden über ein tetraedrisches Intermediat nur erfolgen kann, wenn beide am zentralen sp3-Kohlenstoffatom verbleibende Heteroatome eines ihrer einsamen Elektronenpaare jeweils antiperiplanar zur spaltenden Bindung ausrichten.
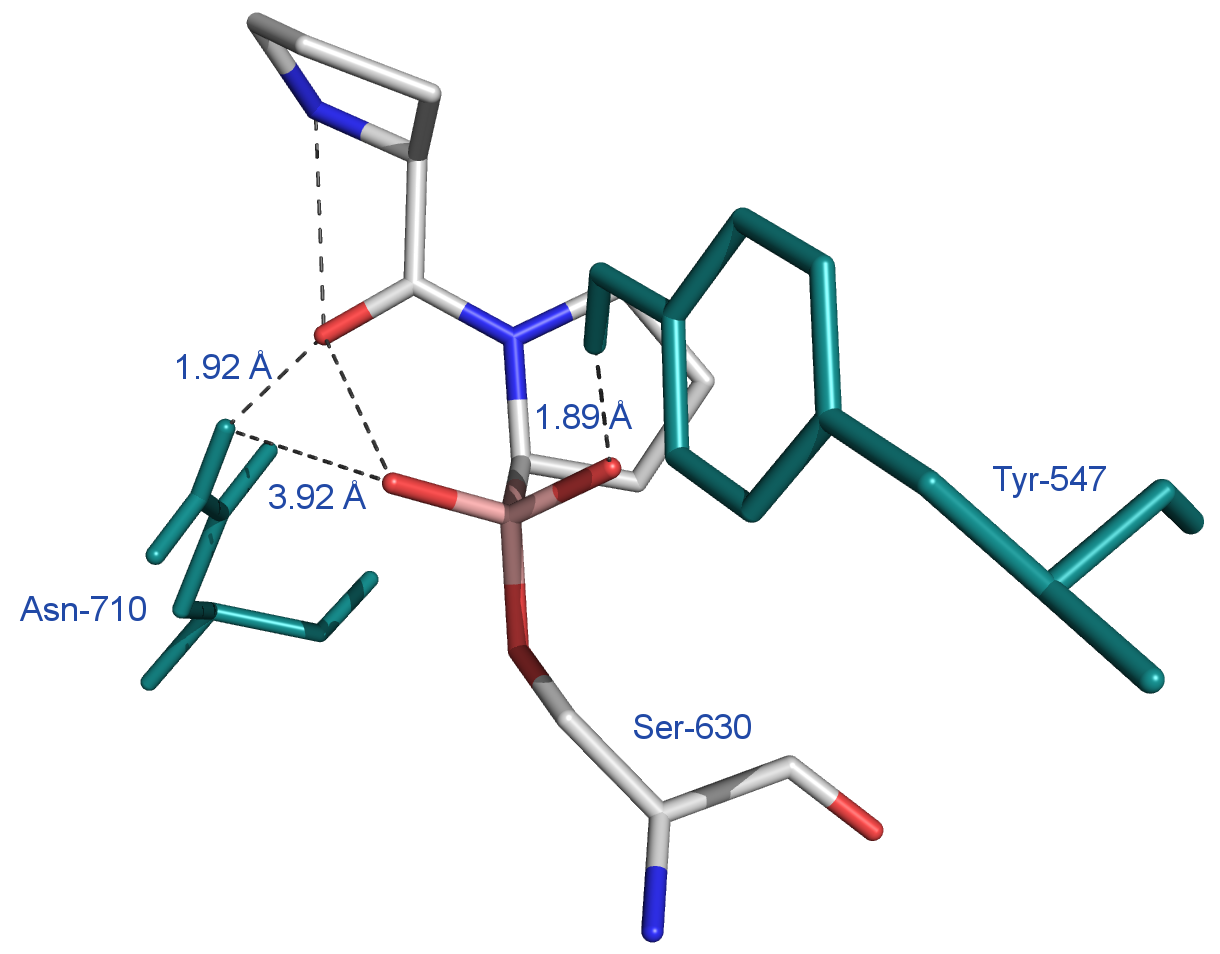
Die Röntgenstrukturanalyse des DPP 4/Pro-Pro-boronsäurekomplexes[12] bestätigt in eindrucksvoller Weise die im Schema 7 angenommene Struktur (Abbildung 4).Außerdem geht aus dem hypothetischen Schema 7 eindeutig hervor, warum die DPP 4 keine N-terminalen Aminosäuren entfernen kann,sondern nur N-terminale Dipeptide abspaltet. Von Interesse ist, dass der Boronsäurekomplex tatsächlich eine tetraedrische Struktur ausbildet, dass eine Wechselwirkung einerseits mit dem Tyr547 des Proteins und einem O-Atom des TI (Abstand = 2,04 A) stattfindet und dass andererseits zwischen dem Asn710 des Proteins und dem Sauerstoffatom der Carbonylgruppe der N-terminalen Peptidbindung eine Wechselwirkung erfolgt (Abstand = 1,93 A). Letztere H-Brücke ist sinnvoll. Sie fördert die Aufrichtung der Carbonylgruppe der N-terminalen Peptidbindung im oben gezeigten Sinne (vergl. Schema 7). Diese Verhältnisse sind in dem Ausschnitt aus der Röntgenkristallstruktur des DPP 4/Boronsäure-Komplexes (Abbildung 4) ersichtlich.
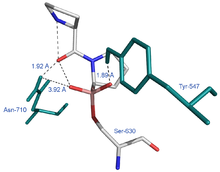 Abb. 4[12]
Abb. 4[12]Ähnliche Bilder zeigen die durch Diprotin A (Abbildung 5 – rechts) und tertBuGly-Pro-Ile (Abbildung 5 – links) erzeugten Strukturen. Hier entstehen asymmetrische tetraedrische Konformationen. Sie stellen sich als antipodisch gegenüber der in dem Schema 7 angenommenen Struktur dar. Sollten diese durch die Röntgenstruktur ermittelten realen antipodischen Strukturen charakteristisch für Transition-State-Inhibitoren sein? Durch diese hypothetische Annahme ließe sich erklären, warum die Verbindungen
- Diprotin A (Ile-Pro-Ile)
Diprotin B (Val-Pro-Leu) und
TertBuGly-Pro-Ile,
die grundsätzlich eine Substratstruktur besitzen, Inhibitoren sind. Der Begriff „hypothetisch“ bringt zum Ausdruck, dass die hier gezeigten Röntgenstrukturen (Abbildung 5) die Konformationen von Inhibitoren darstellen, die Konformationen der TI (TI1) von Substraten können unter den experimentellen Bedingungen der Röntgenkristallstruktruranalyse nicht dargestellt werden.
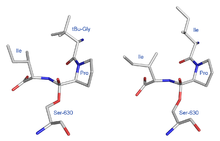 Abb. 5: tBu-GPI Ligand (links) und IPI Ligand (rechts)
Abb. 5: tBu-GPI Ligand (links) und IPI Ligand (rechts)Die Frage: Sind Diprotin A oder Diprotin B Substrate oder Inhibitoren der DPP 4? – wurde im Jahre 1991 von Rahfeld et al. gestellt. Umezawa et al. berichten, dass beide Diprotine als Inhibitoren der DPP 4 wirken. Rahfeld stelle fest, dass sowohl Ile-Pro-Ile als auch Val-Pro-Leu erwartungsgemäß Substrate der DPP 4 sind. Die Autoren sprechen von der Inhibitorwirkung als einem „kinetischen Artefakt“ Die Röntgenkristallstruktur-Studien belegen die Konformation in TI1, die dem Inhibitormolekül zugeordnet werden sollte, denn die Substratstruktur ist mit Sicherheit unter den Kristallisationsbedingungen nicht erfassbar. Um so erstaunlicher ist, dass die Diprotine im wässriger Milieu (kinetische Messungen) nach längerer Zeit (nach ca. 20,5 Stunden unter den in der Literatur angegebenen Bedingungen) vollständig hydrolysiert werden. Offensichtlich stellt sich ein Gleichgewicht der antipodischen TI-Konformationen in verdünnter wässriger Lösung allmählich immer wieder neu ein. Über einen solchen Mechanismus wird der Dualismus bei den Diprotinen klar. Ihre Ki- und KM-Werte sind in der Tabelle 7 gezeigt.
Tabelle 7: Ki- und KM-Werte von Diprotinen Parameter Val-Pro-Leu Ile-Pro-Ile kcat in s−1 27,00 ± 0,05 1,29 ± 0,02 KM in M (1,63 ± 0,10) · 10−5 (3,50 ± 0,30) · 10−6 kcat/KM in M−1·s−1 (1,66 ± 0,10) · 106 (3,69 ± 0,30) · 105 Ki in M (1,88 ± 0,40) · 10−5 (1,28 ± 0,36) · 10−6 Natürliche Substrate der Dipeptidylpeptidase 4
(siehe Dipeptidylpeptidase 4)
Abschließende Bemerkungen
Die Dipeptidylpeptidase 4 ist physiologisch und pharmakologisch von großem Interesse. Einige aktive und spezifische Inhibitoren des Enzyms wurden als potentielle Wirkstoffe gegen Diabetes mellitus Typ II erkannt[13]. Mittlerweile sind die beiden Dipeptidylpeptidase-4-Inhibitoren Sitagliptin und Vildagliptin als Arzneistoffe zur Therapie des Diabetes mellitus Typ 2 zugelassen. Es gibt Hinweise, dass DPP 4 oder verwandte Enzyme eine Rolle bei Prozessen der Wundheilung, möglicherweise in einer Reihe von Krebsarten (Dipeptidyl Peptidase IV und Aminopeptidase N scheinen nach englischsprachigen Veröffentlichungen auch bei der Ausbildung von Thyreoidalcarcinomen involviert zu sein) und in der Pathogenese von AIDS spielen.
Schon vor einigen Jahren wurde die enge Verwandtschaft der Enzyme DP II und PSE (Prolin spezifische Endopeptidase), auch PPCE (Post Proline Cleaving Enzyme) genannt, zur DPP 4 beschrieben. In neuerer Zeit wurde gefunden, dass die DPP 4 Mitglied einer Familie von Dipeptidyl-Peptidasen ist. Es ist zu vermuten, dass auf diesem Gebiet durch vergleichende mechanistische und physiologische Studien noch weitere Einsichten in theoretische und angewandte Fragestellungen resultieren werden.
Einzelnachweise
- ↑ Hopsu-Havu VK, Glenner GG: A new dipeptide naphthylamidase hydrolyzing glycyl-prolyl-beta-naphthylamide. In: Histochemie. 7, Nr. 3, 1966, S. 197–201. PMID 5959122.
- ↑ Drucker DJ: Therapeutic potential of dipeptidyl peptidase IV inhibitors for the treatment of type 2 diabetes. (PDF) In: Expert Opin Investig Drugs. 12, Nr. 1, Januar 2003, S. 87–100. doi:10.1517/13543784.12.1.87. PMID 12517256.
- ↑ Eintrag in der Enzyme Nomenclature des NC-IUBMB
- ↑ Heins J, Welker P, Schönlein C, et al.: Mechanism of proline-specific proteinases: (I) Substrate specificity of dipeptidyl peptidase IV from pig kidney and proline-specific endopeptidase from Flavobacterium meningosepticum. In: Biochim. Biophys. Acta. 954, Nr. 2, Mai 1988, S. 161–9. PMID 2896517.
- ↑ Rahfeld J, Schutkowski M, Faust J, Neubert K, Barth A, Heins J: Extended investigation of the substrate specificity of dipeptidyl peptidase IV from pig kidney. In: Biol. Chem. Hoppe-Seyler. 372, Nr. 5, Mai 1991, S. 313–8. PMID 1678608.
- ↑ Barth,A.:Vier Jahrzehnte im Dienste der Wissenschaft. In:"Acta Facult.Pharm.Univ.Comenianae",52,236-250,2005
- ↑ Schutkowski,M.:"Dissertation an der Universität Halle
- ↑ vergl.:Küllerts,G.,Fischer,G.Barth,A.:Beiträge zum Katalysemechanismus derDipeptidyl Peptidase IV "Acta biol.med. germ.",37,559-567,1978
- ↑ Küllertz,G.,Oehme,P.,Barth,A.,(Hrsg),:Dipeptidyl Peptidase IV - Chemie,Biochemie und physiologische Asdpekte,"Beitr Wirkst.Forsch.,1981.Auch als:Küllertz,G,.Oehme,P.,Barth,A.In:"Pharmazie",36,Nr.7,518 ff,1981
- ↑ Brandt, W., Lehmann, T., Hofmann, T., Schowen, RL., Barth, A.: The probable conformation of substrates recognized by dipeptidyl-peptidase IV and some aspects of the catalytic mechanism derived from theoretical investgations, "J.Comput.Aided Mol.Des.", 6, No.2, 159–174, 1992
- ↑ P.Delongchamps,Stereoelectronic control in the cleavage of tetrahedral intermediates in the hydrolyses of esters and amides,"Tetrahedron" 31,2463-2490,1975
- ↑ a b Engel M, Hoffmann T, Manhart S, et al.: Rigidity and flexibility of dipeptidyl peptidase IV: crystal structures of and docking experiments with DPIV. In: J. Mol. Biol.. 355, Nr. 4, Januar 2006, S. 768–83. doi:10.1016/j.jmb.2005.11.014. PMID 16330047.
- ↑ Barth A.,Thondorf I.,Kovacs P. "Acta Facult.Pharm.Univ.Comenianae",55,5,2008
Literatur
- A. Barth: Der Chemismus der Dipeptidyl-Peptidase-IV – ein Überblick. In: Digitale Bibliothek Sachsen-Anhalt. MLU Halle-Wittenberg, Mathematisch-Naturwissenschftlich-Technische Fakultät, 22. April 2010, abgerufen am 19. April 2011.
- A. Barth, I. Thondorf, J. Stano: Gedanken zum Mechanismus der Serinproteasen,Teil I – Die Funktion der Tetrade: Asp….His….Ser….Xaa. In: Acta Facult. Pharm. Univ. Comenianae 51, 15–26 (2004).
- A. Barth, I. Thondorf, S. Gebauer, W. Brandt, K. Neubert, J. Stano, M. Psenak, P.Kovacs: Gedanken zum Mechanismus der Serinproteasen, Teil II–Dipeptidyl-Peptidase IV (DP IV/CD 26). In: Acta Facult. Pharm. Univ. Comenianae 52, 7–21 (2005).
- A. Barth, I. Thondorf, S. Gebauer, K. Neubert, P. Kovacs, J. Stano: Gedanken zum Mechanismus der Serinproteasen, Teil III – a) Das tetraedrische Intermediat im Acylierungsprozess. b) Die Boronsäuren. In: Acta Facult. Pharm. Univ. Comemianae 53, 5–15 (2006)
- A. Barth, I. Thondorf, K. Neubert, S. Hoffmann, P. Kovacs, J. Stano: Gedanken zum Mechanismus der Serinproteasen, Teil IV – Trasition State Inhibitoren – Das asymmetrische tetraedrische Intermediat TI 1. In: Acta Facult. Pharm. Univ. Comenianae 53, 16–21 (2006).
- A. Barth, I. Thondorf: "Gedanken zum Mechanismus der Serinproteasen, Teil V - Dipeptidyl Peptidase IV -Biochemisch-physiolögische Aspekte" In: "Acta Facult." Pharm. Univ. Comenianae 54, 7 - 13 (2007).
- A. Barth, I. Thondorf, P. Kovacs: "Gedanken zum Mechanismus der Serinproteasen, Teil VI - DP IV Inhibitoren. Wirkstoffe gegen Diabetes Typ II mit neuartigem Wirkmechanismus" In: "Acta Facult." Pharm. Univ. Comenianae 55, 5 - 10 (2008)
- A. Barth: "Gedanken zum Mechanismus der Serinproteasen, Teil VII - Ein Überblick [1966 - 2006]" In: "Acta Facult." Pharm. Univ. Comenianae 55, 11 - 22 (2008)
- S. Ansorge (Hrsg.): Abstracts of the 2nd International Conference on Dipeptidyl Aminopeptidases – Basic Sciences and Clinical Applications. Magdeburg, Germany 2005.
- U. Langdeckel, D. Reinhold, U. Bank (Hrsg.): Dipeptidyl Aminopeptidases – Basic Science and Clinical Applications (Proceedings) in Advances in Experimental Medicine and Biology. Vol. 575, Springer 2006.
- W. Brandt, T. Hofmann: The Recognition Conformation of Substances of the Dipeptidyl Peptidase IV. In: Biol. Zentr. bl. 107, 21–30 (1978).
- G. Küllertz, G. Fischer, A. Barth: Beiträge zum Katalysemechanismus der Dipeptidyl-Peptidase IV. In: Acta biol. med. germ. 37, 559–567 (1978).
- K. Ludwig, Y. Shuling, H. Fan, W. Reutter, Ch. Böttcher: The 3D stucture of DPP IV/CD 26 as obtained by cryo-TEM and single partial Analysis. In: Biochem. Biophys. Res. Commun. 304, 73–77 (2003).
- M. Engel, T. Hofmann, S. Manhart, U. Heiser, S. Chambre, R: Huber, H.-U. Demuth, W. Bode: Rigitity and Flexibility of Dipeptidyl Peptidase IV: Crystal Structures of and Docking Experiments with DP IV. In: J. Mol. Biol. 355 768–783 (2006).
- R. Thoma, B. Löffler, M. Stihle, M. Huber, W. Ruff, A. und M. Hennig: Structural basis of proline-specific exopeptidase activity as observed in Human dipeptidyl peptidase-IV. In: Structure (Camb). 11, 947–959 (2003).
- J. Rahfeld, M. Schierhorn, B. Hartrodt, K. Neubert, J. Heins: Are diprotin A (Ile-Pro-Ile) and diprotin B (Val-Pro-Leu) inhibitors or substrates of dipeptidyl peptidase IV? In: Biochim. Biophys. Acta. 1976, 314–316 (1991).
- H. Umezawa, T. Aoyagi, H. Naganawa, M. Hamada T. Takeuchi: Diprotins A and B, Inhibitors of Dipeptidyl Aminopeptidase IV, Produced by Bacteria. In: J. Antibiotics. 37, 422–425 (1984).







Wikimedia Foundation.