- Pick-Krankheit
-
Klassifikation nach ICD-10 G31.0 Umschriebene Hirnatrophie
– Pick-KrankheitF02.0* Demenz bei Pick-Krankheit ICD-10 online (WHO-Version 2011) Bei der Pick-Krankheit (oder Frontotemporale Demenz, früher auch Picksche Krankheit) handelt es sich um eine meist vor dem 60. Lebensjahr auftretende neurodegenerative Erkrankung im Stirn- bzw. Schläfenlappen des Gehirns. Bei dem Krankheitsbild handelt es sich um eine Form der frontotemporalen Demenz (FTD). Bei dieser Erkrankung steht zunächst nicht die Beeinträchtigung von Gedächtnisleistungen im Vordergrund sondern eine fortschreitende Veränderung der Persönlichkeit und der sozialen Verhaltensweisen.[1]
Als Demenz ist sie seltener als die Alzheimersche Krankheit bzw. DVAT, welche ebenfalls zur Gruppe der neurodegenerativen Erkrankungen gezählt wird. Ein familiäres Auftreten wird bei Morbus Pick häufiger beobachtet (in etwa vierzig Prozent der Krankheitsfälle, Angaben zur familiären Häufung schwanken). Ein autosomal-dominanter Erbgang lässt sich in weniger als zehn Prozent der Fälle feststellen.
Die Abgrenzung zum Begriff des "Frontalhirnsyndroms" (einer Beschreibung von Folgen einer lokalisierten Störung im Gehirn, keinem eigenständigen Krankheitsbild) wird im entsprechendem Artikel geleistet. Die Erkrankung darf auch nicht mit dem Morbus Niemann-Pick, einer Sphingolipidose verwechselt werden.
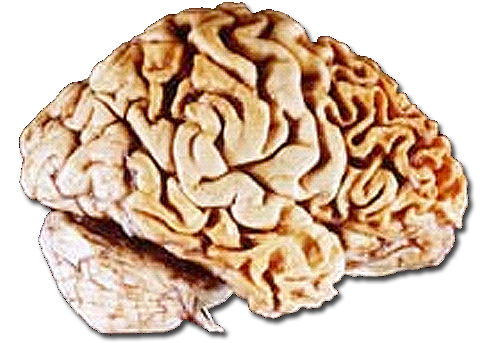
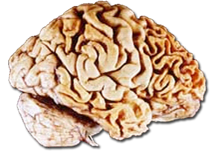 Frontotemporale Degeneration
Frontotemporale Degeneration
Inhaltsverzeichnis
Historisches
Erstmals beschrieben wurde Morbus Pick um 1900 von dem Prager Neurologen Arnold Pick, der bei der Obduktion früh verstorbener, damals so genannter "Schwachsinniger" eine Atrophie (Gewebsschwund) der Stirn- und Schläfengehirnlappen feststellte und dies als besondere Krankheit einstufte. In den 1920er Jahren wurden weitere Fälle beschrieben und deswegen wurde das Bild nach dem Erstbeschreiber als Picksche Krankheit oder Morbus Pick bezeichnet.
Ursache der Krankheit
Welche Faktoren genau das Auftreten der Krankheit verursachen, ist noch weitgehend unerforscht. Als einer der Auslöser konnte das für das Tau-Protein codierende MAPT-Gen identifiziert werden. Eine andere auslösende Mutation im für Präsenilin-1 codierenden PSEN1-Gen ist ebenfalls belegt.[2][3] Die pathologische Anhäufung eines Proteins namens TDP-43 in den Nervenzellen ist in rund der Hälfte der Fälle belegt. Diese Fälle treten familiär gehäuft auf. Außerdem wurden Mutationen in im TMEM106B-Gen gefunden, welche die Krankheit als starker Risikofaktor begünstigen (Chromosom 7p21). Eine Assoziation mit der Anhäufung von TDP-43 wird vermutet, ist bisher aber noch nicht belegt.[4] Als einziges gesichertes morphologisches Merkmal können nach dem Tod des Patienten charakteristische Einschlusskörper (in diesem Fall „Pick-Körpern“ genannt) dargestellt werden, die 1911 von Alois Alzheimer bei frontotemporalen Demenzen beschrieben wurden (Tauopathie).
Epidemiologie
Die Prävalenz wird mit 3,4/100.000 angenommen. Die Krankheit beginnt meist im 50. bis 60. Lebensjahr, wobei die Spanne sehr groß ist (20-85 Jahre). Frauen sind von der Pickschen Krankheit etwa gleich häufig betroffen wie Männer. Morbus Pick stellt mit einer Häufigkeit von 3 - 9 Prozent (je nach Quelle) unter allen Demenzen einen seltenen Typus dar. Geht man von etwa 1.300.000 Demenzkranken in Deutschland aus[5], wären schätzungsweise mindestens 40.000 Menschen von der Pickschen Krankheit betroffen.[6] Die mittlere Überlebenszeit liegt bei 8 Jahren. Es sollte aber berücksichtigt werden, dass das Auftreten der ersten Symptome im Rückblick oft schwierig zu bestimmen ist und alle Schätzungen zur Dauer des Krankheitsverlaufs ungenau sein dürften.[7][8]
Symptome und Krankheitsverlauf
Morbus-Pick-Patienten leiden im Allgemeinen an Verhaltensauffälligkeiten, Persönlichkeitsveränderungen, Sprach- und Gedächtnisstörungen und dem Verlust anerzogener Verhaltensregeln. Der Verlauf der Krankheit ist eher langsam fortschreitend. Üblicherweise gehen die beiden zuerst genannten Symptome einer Störung des Gedächtnisses voraus.
Klinisch sind zwei Symptomkomplexe zu beobachten – entweder „passiv“ anmutende Zeichen wie:
- Apathie
- Antriebslosigkeit
- Vernachlässigung der Körperpflege
- affektive Verflachung
- Verwahrlosung
oder gegensätzlich wirkende Symptome wie:
- Triebhaftigkeit
- Euphorie
- Verlust ethischer Werte (Kriminalität)
- generelle Enthemmungsphänomene (Esssucht, Vergröberung der Tischmanieren, Witzelsucht und sexuelle Anzüglichkeiten/Handlungen)
Im weiteren Verlauf der Erkrankung kommen Sprach- und Orientierungsstörungen hinzu, bis sich schließlich das Vollbild der frontotemporalen Demenz mit Muskelversteifung (Rigor), Pflegebedürftigkeit sowie Harn- bzw. Stuhlinkontinenz ausgebildet hat.
Je nach betroffener Hirnregion ist das Auftreten von Unterformen der frontotemporalen Demenz wie beispielsweise der progredienten (fortschreitenden) Aphasie oder der semantischen Demenz möglich.
Diagnose
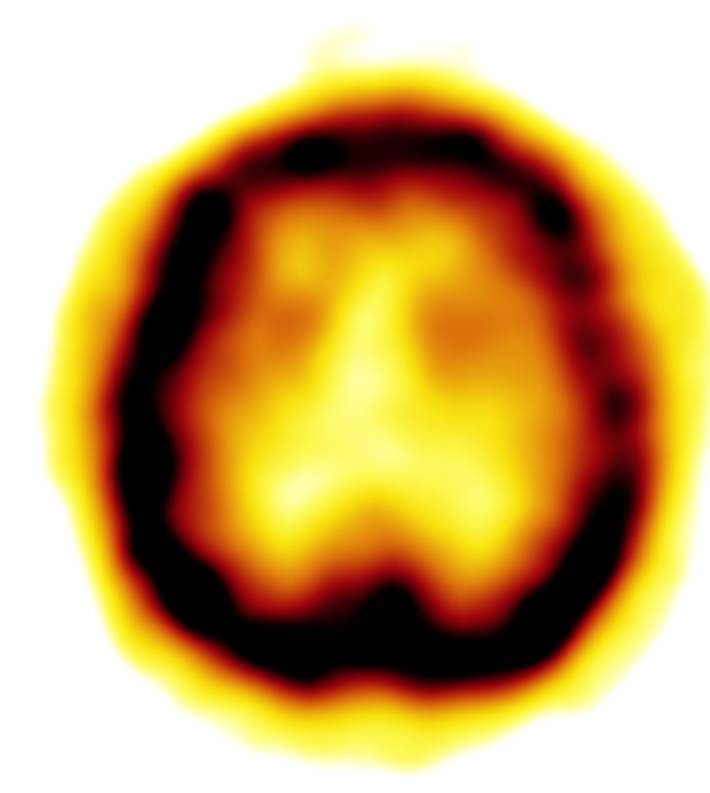
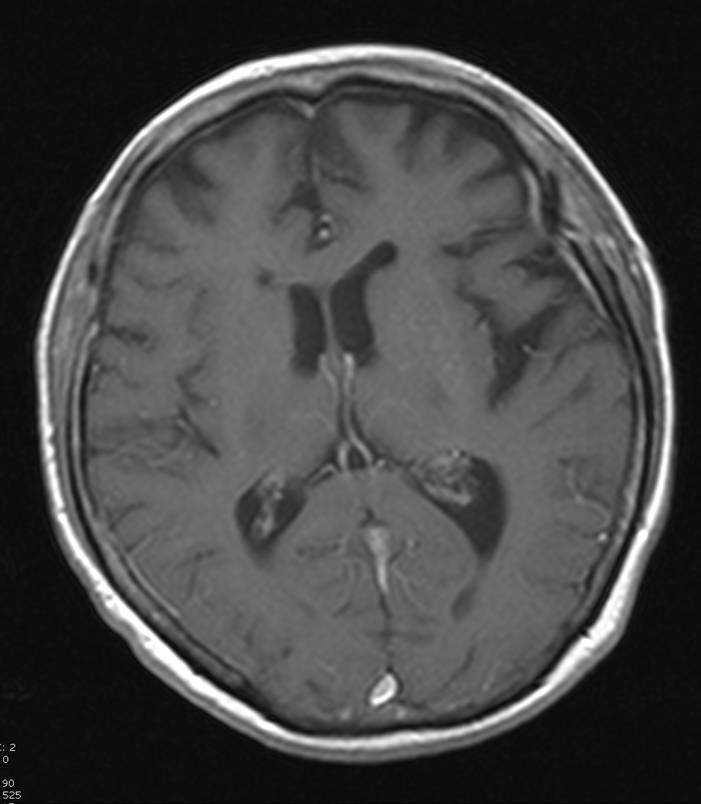
 Hirn-PET mit vermindertem Glucose-Stoffwechsel im linken Temporallappen.
Hirn-PET mit vermindertem Glucose-Stoffwechsel im linken Temporallappen.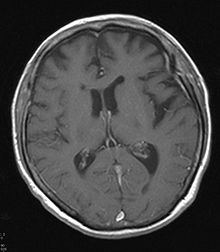 Zugehörige MRT mit entsprechender Atrophie.
Zugehörige MRT mit entsprechender Atrophie.Neben klinischen Verdachtsmustern kann in vivo eine nuklearmedizinische Diagnostik mit Darstellung der Gehirnperfusion (mit 99mTc-HMPAO) (SPECT) oder des Glucose-Stoffwechsels des Gehirns mit 18FDG (PET) weiterhelfen - typischerweise sind in den Gehirnabschnitten, die dem Krankheitsnamen (frontotemporal) entsprechen, die Durchblutung und der Glucose-Stoffwechsel herabgesetzt.
Eine sichere Diagnose dieser seltenen Demenzform ist erst nach dem Tod möglich. Die Krankheit lässt sich jedoch anhand der Symptomkonstellation vermuten und durch Abklärung möglicher Ausschlussdiagnosen eingrenzen.
Unter dem Mikroskop finden sich im Bereich des Schläfenlappens (Temporallappens) und des Stirnlappens (Frontallappens) degenerative Veränderungen („Nervenzelluntergänge“), die durch Ganglienzellenschwund und Gliose unterhalb der Hirnrinde verursacht wurden. Unklar ist die Rolle der so genannten „Pickschen Körper“ (kugelförmige Anhäufungen des τ-Proteins) in den Nervenzellen des Frontal- bzw. Temporalhirns. Es lässt sich jedenfalls kein linearer Zusammenhang zwischen der Menge an Pickschen Körpern und dem Auftreten der Demenz herstellen; auch bei nicht an Morbus Pick erkrankten Patienten fanden sich Picksche Körper.
Einteilung nach Verlaufsform
Klinisch lässt sich diese Demenz oft in 3 Prägnanztypen unterteilen, die vor allem im Frühstadium unterscheidbar sind. Sie gehen später im Verlauf ineinander über:
- Frontale/frontotemporale Verlaufsform mit führender Wesensänderung (Haupttyp)
- Primär-progressive Aphasie (also eine führende nichtflüssige Aphasie' (Sprachstörung) und evtl. linkstemporale Atrophie)
- Semantische Demenz (führende bitemporale Atrophie, Defizit des Wissens über Wort- und Objektbedeutungen, fehlerhafte syntaktische Sprachverarbeitung, Defizit des „Weltwissens“ zu allgemeinen Fakten, visuell-gnostische Störungen).
Therapie
Weder eine Heilung noch eine spezifische medikamentöse Therapie sind derzeit möglich oder nur in Aussicht (Stand: 11/2006). Die gegen Morbus Alzheimer zeitweise wirksamen Acetylcholinesterasehemmer erwiesen sich bei der Behandlung frontotemporaler Demenzen als wirkungslos. Psychotische Begleiterscheinungen der Pickschen Krankheit können allerdings durch die Gabe von Neuroleptika (etwa Pipamperon und Levomepromazin) gelindert werden.
Im fortschreitenden Verlauf ist eine Dauerhospitalisierung von Pick-Patienten mit intensiver Pflege erforderlich. Psychologische/psychotherapeutische Unterstützung der Angehörigen ist angesichts der schwerwiegenden Symptomatik unumgänglich.
Literatur
- A. Brun et al.: Clinical and neuropathological criteria for frontotemporal dementia.. In: J Neurol Neurosurg Psychiatry. 57 (1994), S. 416–418
- J. Diehl: Frontotemporale Demenz. In: Geriatrie-Journal. Der ältere Patient in Praxis und Klinik 4 (2002), S. S. 36–37
- J. Diehl, A. Kurz: Frontotemporale Demenz. In: Wiener Medizinische Wochenschrift, 152 (2002), S. 92–97
- J. Diehl, I. R. Mackenzie, H. Förstl, A. Kurz: Die frontotemporale Demenz: Ergebnisse der „Frontotemporal Dementia & Pick's Disease Conference“. In: Nervenarzt, 74 (2003), S. 785–788
Weblinks
- [2] Deutsche Alzheimer Gesellschaft e.V., Selbsthilfe Demenz, Links zu Angehörigengruppen, Internetforen u.a.
- [3]Deutsche Alzheimer Gesellschaft, Die Frontotemporale Demenz (Pick-Krankheit), Informationsblatt 2003
- Erlebnisbericht einer Angehörigen SPIEGEL online
- Informationen zur frontotemporalen Demenz auf Wegweiser-Demenz, dem Portal des Bundesministeriums für Familie, Senioren, Frauen und Jugend
Einzelnachweise
- ↑ J. Greck, N. Lautenschlager, A. Kurz: Klinische Aspekte der frontotemporalen Demenz. Fortschr. Neurol. Psychiatr. 2000 Oct; 68(10): S. 447-457.
- ↑ Frontotemporale Demenz bei Online Mendelian Inheritance in Man.
- ↑ Pick-Krankheit bei Online Mendelian Inheritance in Man.
- ↑ Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS et alia : Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions., Nat Genet. 2010 Feb 14. PMID 20154673
- ↑ http://www.wegweiser-demenz.de/gesellschaft-und-demenz.html Abgerufen am 4. Dezember 2010
- ↑ [1]Die Frontotemporale Demenz (Pick-Krankheit), Deutsche Alzheimer Gesellschaft, September 2003
- ↑ J. Diehl, I. R. Mackenzie, H. Förstl, A. Kurz: Die frontotemporale Demenz: Ergebnisse der „Frontotemporal Dementia & Pick's Disease Conference“. Nervenarzt. 2003, 74: 785–788.
- ↑ J. L. Cummings. The Neuropsychiatry of AD and Related Dementias. Taylor & Francis, London, 2003

Bitte den Hinweis zu Gesundheitsthemen beachten!
Wikimedia Foundation.