- Proteinfehlfaltungserkrankung
-
Als Proteinfehlfaltungserkrankungen, auch Proteinfaltungserkrankungen (engl. protein misfolding diseases oder protein misfolding disorders oder conformational diseases) genannt, bezeichnet man solche Erkrankungen, die durch falsche gefaltete Proteine verursacht werden. Die fehlgefalteten Proteine werden entweder in den Zellen eingelagert oder im Proteasom abgebaut. Im ersten Fall bilden sich dabei toxische Ablagerungen (Plaques), im zweiten tritt ein Funktionsverlust, bedingt durch einen Mangel des entsprechenden Proteins in der Zelle beziehungsweise im gesamten Organismus, ein. Beides kann über die Zeit für den Betroffenen pathologisch werden und abhängig vom betroffenen Protein zu unterschiedlichen Erkrankungen führen.
Inhaltsverzeichnis
Beschreibung
 Schematische Darstellung der Proteinqualitätskontrolle im Endoplasmatischen Retikulum.
Schematische Darstellung der Proteinqualitätskontrolle im Endoplasmatischen Retikulum.
Das über einen Transportprotein in das ER eingeschleuste lineare ungefaltete Protein beginnt sich mit der Hilfe von Chaperonen (nicht eingezeichnet) zu falten. Wird es von der Proteinqualitätskontrolle als korrekt gefaltet erkannt, wird es per Vesikel aus dem ER ausgeschleust. Fehlgefaltete Proteine werden über ein Transportprotein in das Zytosol geschleust und dort in einem Proteasom in Fragmente zerlegt. Amorphe Aggregate können auch über Autophagozytose abgebaut werden.
Werden zu viele Moleküle wegen Fehlfaltung abgebaut, so kann dies zu einem Funktionsverlust in der Zelle oder dem ganzen Organismus führen. Bilden sich zu viele unlösliche, nicht mehr abbaubare Aggregate, so entstehen für die Zelle und den gesamten Organismus toxische Ablagerungen.[1]In den meisten Zellen aller Organismen werden im Rahmen der Proteinbiosynthese ständig die verschiedensten Proteine (Eiweiße) produziert, die in der Zelle und im gesamten Organismus die unterschiedlichsten Funktionen erfüllen. Für eine korrekte Funktion eines Proteins ist dessen Tertiärstruktur von entscheidender Bedeutung. Diese Struktur wird durch einen Prozess erreicht, der Proteinfaltung genannt wird. Die Proteinfaltung ist ein komplexer und empfindlicher Vorgang. Die korrekte Proteinfaltung wird von der Proteinqualitätskontrolle überwacht. Statistisch gesehen werden etwa 30 % aller Proteine aus der Proteinbiosynthese nicht korrekt gefaltet und normalerweise innerhalb von etwa zehn Minuten im Proteasom der Zelle abgebaut.[2][3]
Die Gründe für eine falsche Proteinfaltung sind vielschichtig. Genmutationen in Exons, die zu Veränderungen in der Aminosäuresequenz, also der Primärstruktur des Genproduktes führen, haben unmittelbare Einflüsse auf die Sekundär- und Tertiärstruktur, beziehungsweise auf die Proteinfaltungskinetik. Auch Fehler bei der Transkription oder der Translation können zu Fehlfaltungen der Proteine führen. Die fehlerhaften Proteine werden auch als defekte ribosomale Produkte (engl. defective ribosomal products, DRiPs) bezeichnet.[4]
Gain-of-toxic-function
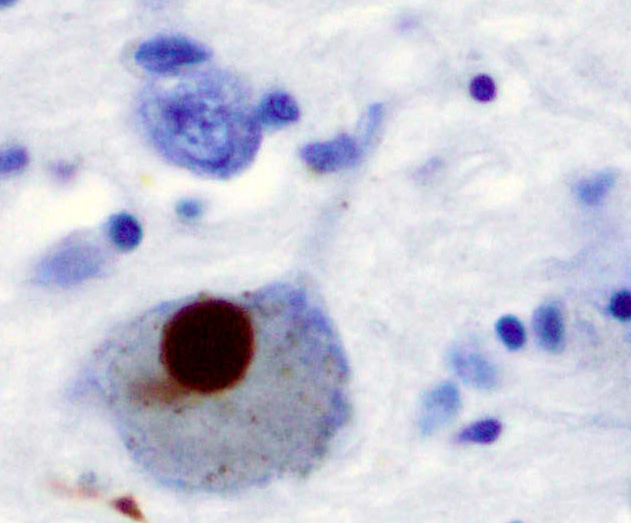
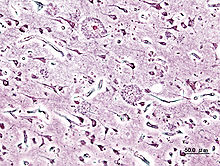 Feingeweblicher Schnitt mit Alzheimer-Plaques
Feingeweblicher Schnitt mit Alzheimer-Plaques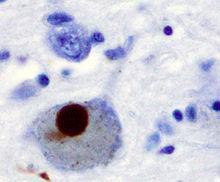
Können die DRiPs im Proteasom nicht abgebaut werden, beispielsweise, weil sie sich zuvor zu Aggregaten zusammengelagert haben, so sammeln sich die DRiPs in der Zelle an. Dort können sie mit der Zeit pathologisch werden, das heißt zu spezifischen Erkrankungen führen. Die Proteinaggregate führen vor allem zu neurodegenerativen Erkrankungen wie Parkinson-Krankheit, Alzheimer-Krankheit oder Chorea Huntington. Die Aggregate haben in den Zellen eine neue toxische Funktion. Für die toxische Wirkung innerhalb der Zellen wird der englischsprachige Begriff gain of (toxic) function verwendet.[5]
In der englischsprachigen Fachliteratur haben sich für diese Form der Proteinfehlfaltungserkrankungen die Begriffe proteinopathy und proteopathy etabliert. Die dem entsprechenden deutschen Begriffe Proteopathie und Proteinopathie (der Präfix Proteo- = ‚Protein‘ und der Suffix -pathie = ‚Erkrankung‘) haben sich dagegen in der deutschsprachigen Fachliteratur bisher kaum durchgesetzt.
Derzeit (Stand 2011) sind über 100 Proteinopathien bei Mensch und Tier bekannt. Sie werden durch die Ablagerung von etwa 20 nicht-homologen Proteine verursacht. Eine große und wichtige Gruppe bilden dabei die Amyloidosen.[6]
Zu den Proteinfehlfaltungserkrankungen mit gain of toxic function zählen unter anderem folgende Erkrankungen:
Erkrankung verursachendes Protein Anmerkungen Alzheimer-Krankheit[7] β-Amyloid, Tau Typ Tauopathie Pick-Krankheit Tau Typ Tauopathie Kortikobasale Degeneration Tau Typ Tauopathie Silberkornkrankheit Tau Typ Tauopathie Progressive supranukleäre Blickparese Tau Typ Tauopathie Parkinson-Krankheit[7] α-Synuclein Typ Synucleinopathie Multisystematrophie α-Synuclein Typ Synucleinopathie Lewy-Körper-Demenz α-Synuclein Typ Synucleinopathie Chorea Huntington[7] Huntingtin Polyglutaminerkrankung (polyglutamine expansion disease) aus der Familie der Trinukleotiderkrankungen Spinozerebelläre Ataxie[8] u.a. Ataxin-2 Polyglutaminerkrankung Spinobulbäre Muskelatrophie Typ Kennedy[9] Androgenrezeptor Polyglutaminerkrankung Dentatorubro-Pallidoluysische Atrophie (DRPLA) Atrophin Polyglutaminerkrankung ATTR-Amyloidose und AP-Amyloidose Transthyretin Amyloidose erbliche systemische Amyloidose (meist) Transthyretin Amyloidose nicht-erbliche systemische Amyloidose[10] Immunglobulin-Leichtketten (AL-Typ), APP-Fragmente (AA-Typ), β2-Mikroglobulin (AB-Typ) Amyloidose (siehe auch: β2-Mikroglobulin-assoziierte Amyloidose (Hämodialyse-Amyloidose)) Transmissible spongiforme Enzephalopathien[11] Prionen Prionenerkrankungen u.a. Creutzfeldt-Jakob-Krankheit, tödliche familiäre Schlaflosigkeit, Gerstmann-Sträussler-Scheinker-Syndrom, Kuru Katarakt (grauer Star)[12] multiple Proteine Denaturierung von verschiedenen Linsen-Proteinen Amyotrophe Lateralsklerose (zumindest bei einigen Varianten der Erkrankung)[13] Superoxiddismutase Alexander-Krankheit[14] Saures Gliafaserprotein (GFAP) CADASIL[15] Notch 3 Sichelzellenanämie Hämoglobin Frontotemporallappen-Degeneration (FTLD-TLP) TDP-43 Alveolarproteinose Surfactant-Protein-C Sporadische Einschlusskörpermyositis[16] β-Amyloid (noch nicht gesichert) Diabetes mellitus Typ 2[17] Amylin Loss-of-physiological-function
Zu den Proteinfehlfaltungserkrankungen zählen außerdem die Erkrankungen, bei denen die fehlgefalteten Proteine im Proteasom zerlegt werden, wodurch keine ausreichenden Mengen des Proteins den Zellen beziehungsweise dem Organismus zur Verfügung stehen.[18] Dieser Funktionsverlust, engl. loss of (physiological) function, kann zu Erkrankungen wie beispielsweise Mukoviszidose führen.[19] Bei den meisten Patienten mit Mukoviszidose liegt eine ΔF508-Mutation (Typ Deletion) im CFTR-Protein – einem Chloridkanal – vor. Die Deletion von drei Nukleotiden bewirkt, dass an Position 508 von CFTR die Aminosäure Phenylalanin (im Einbuchstabencode F) fehlt. Durch diese Mutation wird das hochkomplexe CFTR, das unter anderem 21 transmembrane Proteindomänen aufweist, in seiner Faltungskinetik stark verändert. Der Faltungsprozess des CFTR-Wildtyps benötigt bereits über zwei Stunden und lediglich etwa 30 % der synthetisierten CFTR-Moleküle faltet schnell genug, um der ER-assoziierten Proteindegradation (ERAD) zu entkommen. Das ΔF508-CFTR faltet noch etwas schlechter und wird komplett abgebaut, obwohl es im Prinzip als Ionenkanal voll funktionsfähig wäre. Den von dieser Mutation betroffenen Patienten fehlt der Chloridkanal (=Funktionsverlust), was zur folge hat, dass die Zusammensetzung der Sekrete verschiedener exkretorische Drüsen drastisch verändern.[20][21]
Ein Verlust an physiologischer Funktion liegt unter anderem bei folgenden Erkrankungen vor:
Erkrankung defektes Protein/Gen Anmerkungen Zystennieren[22] Polycystin-1 Morbus Charcot-Marie-Tooth[23] Aminoacyl-tRNA-Synthetase (AARS) X-chromosomales lymphoproliferatives Syndrom[24] SH2D1A Morbus Hirschsprung[25] Rezeptor-Tyrosinkinase Ret Homocystinurie und Methylmalonazidurie[26] MMACHC Patellahypoplasie[27] TBX4 Sklerosteose[28] Sclerostin Mukoviszidose[29][30] CFTR Phenylketonurie[31] Phenylalaninhydroxylase Hand-Fuß-Genital-Syndrom[32] Homeobox-Protein A13 lysosomale Speicherkrankheiten[33] verschiedene lysosomale Enzyme über 40 einzelne Erkrankungen, u.a. Morbus Gaucher[34], Morbus Fabry[35] Tay-Sachs-Syndrom[36] oder Morbus Krabbe[37] QT-Syndrom[38] u.a. hERG Angelman-Syndrom UBE3A erblicher Brustkrebs[39] BRCA1 Gain-of-funtion und Loss-of-function
Darüber hinaus gibt es Proteinfehlfaltungserkrankungen, bei denen sowohl ein Funktionsverlusts, als auch die toxischen Proteinablagerungen pathologisch werden können. Ein Beispiel hierfür ist der Alpha-1-Antitrypsin-Mangel. Eine Mutation im SERPINA1-Gen, das für das Akute-Phase-Protein α-1-Antitrypsin – ein Proteaseinhibitor – kodiert, bewirkt eine Fehlfaltung von α-1-Antitrypsin. α-1-Antitrypsin wird im Wesentlichen von Hepatozyten in der Leber exprimiert. Wegen der Fehlfaltung kann es nicht von den Heptozyten sezerniert werden und es bildet intrazelluläre Ablagerungen. Der Funktionsverlust führt bei den betroffenen Patienten zu einem progredienten Lungenemphysem, da durch den Mangel an α-1-Antitrypsin das Enzym Leukozytenelastase (engl. human leukocyte elastase, HLE) ungebremst das Lungengerüst zerstören kann. Die Ablagerungen von α-1-Antitrypsin in den Hepatozyten führen parallel zum Lungenemphysem zu einer Leberzirrhose.[19][40][41]
Behandlungskonzepte
 Saproterin, ein pharmakologisches Chaperon
Saproterin, ein pharmakologisches Chaperon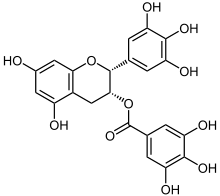 Epigallocatechingallat, ein Bestandteil des grünen Tees, unterstützt den Vorgang der Proteinfaltung
Epigallocatechingallat, ein Bestandteil des grünen Tees, unterstützt den Vorgang der ProteinfaltungDie Proteinfehlfaltungerkrankungen sind derzeit nicht heilbar. Für die häufigsten neurodegenerativen Erkrankungen die durch ein gain of toxic function verursacht werden, gibt es noch keine kausale oder kurative Therapien. Die Behandlung der Patienten erfolgt meist symptomatisch oder rein palliativ. Es gibt einige zukünftige kurative Behandlungskonzepte, wie beispielsweise die Gentherapie, die aber noch viele Jahre von einer Zulassung entfernt sind.
Proteinfehlfaltungserkrankungen, die durch einen Funktionsverlust des Proteins hervorgerufen werden, sind teilweise kurativ behandelbar. Bei der Enzymersatztherapie wird den Patienten das fehlende Protein, das gentechnisch produziert wird, mittels Infusion künstlich zugeführt. Chaperon-Therapien können für beide Arten von Proteinfehlfaltungserkrankungen zukünftige Behandlungsmöglichkeiten sein.[42] Molekulare Chaperone sind Proteine, deren wichtigste Aufgabe es ist, neu synthetisierten Proteinen bei ihrer korrekten Faltung zu „helfen“. Darüber hinaus wurden „künstliche“ chemische und pharmakologische Chaperone identifiziert und entwickelt, die den Faltungsprozess ebenfalls unterstützen.[43] Der Wirkstoff Sapropterin zur Behandlung der Phenylketonurie ist ein Beispiel für ein zugelassenes pharmakologisches Chaperon. Der Iminozucker 1-Deoxygalactonojirimycin (DGJ), internationaler Freiname Migalastat ist ein anderes pharmakologisches Chaperon, das derzeit (Stand Oktober 2011) in der klinischen Phase III zur Erprobung der Wirksamkeit bei Patienten mit Morbus Fabry ist.[44]
Das vor allem in grünem Tee vorkommende Epigallocatechingallat (EGCG) ist offensichtlich in der Lage die korrekte Faltung von Proteinen zu unterstützen.[45] Bei In-vitro-Versuchen konnte EGCG die Fibrillogenese (die Bildung von Fibrillen) von Huntingtin,[46] α-Synuclein und β-Amyloid inhibieren.[47][48][49] EGCG sorgt dafür, dass statt der faserförmigen toxischen Fibrillen ungefährliche sphärische Oligomere entstehen. Offensichtlich ist es auch in der Lage bereits gebildete Plaques aufzulösen.[47] In Farbmäusen konnte die Plaque-Belastung im Kortex, Hippocampus und im entorhinalen Kortex um jeweils etwa 50 % gesenkt werden.[50]
Weiterführende Literatur
Fachbücher
- M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9 Eingeschränkte Vorschau in der Google Buchsuche
- J. Ovádi, F. Orosz (Hrsg.): Protein Folding and Misfolding: Neurodegenerative Diseases. Verlag Springer, 2009, ISBN 1-402-09433-7 Eingeschränkte Vorschau in der Google Buchsuche
- H. J. Smith, C. Simons, R. D. E. Sewell: Protein misfolding in neurodegenerative diseases. CRC Press, 2008, ISBN 0-849-37310-7
- V. N. Uversky, A. L. Fink (Hrsg.): Protein misfolding, aggregation and conformational diseases. Verlag Springer, 2007, ISBN 0-387-36529-X Eingeschränkte Vorschau in der Google Buchsuche
- R. M. Murphy, A. M. Tsai: Misbehaving proteins – protein (mis)folding, aggregation, and stability. Verlag Springer, 2006, ISBN 0-387-30508-4 Eingeschränkte Vorschau in der Google Buchsuche
- P. Bross, N. Gregersen (Hrsg.): Protein misfolding and disease: principles and protocols. Humana Press, 2003, ISBN 1-588-29065-4 Eingeschränkte Vorschau in der Google Buchsuche
Review-Artikel
- K. F. Winklhofer, J. Tatzelt, C. Haass: The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. In: The EMBO journal. Band 27, Nummer 2, Januar 2008, S. 336–349, ISSN 1460-2075. doi:10.1038/sj.emboj.7601930. PMID 18216876. PMC 2234348.
- H. Naiki, Y. Nagai: Molecular pathogenesis of protein misfolding diseases: pathological molecular environments versus quality control systems against misfolded proteins. In: Journal of biochemistry. Band 146, Nummer 6, Dezember 2009, S. 751–756, ISSN 1756-2651. doi:10.1093/jb/mvp119. PMID 19643812.
- L. M. Luheshi, C. M. Dobson: Bridging the gap: from protein misfolding to protein misfolding diseases. In: FEBS letters. Band 583, Nummer 16, August 2009, S. 2581–2586, ISSN 1873-3468. doi:10.1016/j.febslet.2009.06.030. PMID 19545568.
- M. Stoppini, L. Obici u.a.: Proteomics in protein misfolding diseases. In: Clinical chemistry and laboratory medicine. Band 47, Nummer 6, 2009, S. 627–635, ISSN 1434-6621. doi:10.1515/CCLM.2009.164. PMID 19527136.
- H. Ecroyd, J. A. Carver: Unraveling the mysteries of protein folding and misfolding. In: IUBMB life. Band 60, Nummer 12, Dezember 2008, S. 769–774, ISSN 1521-6551. doi:10.1002/iub.117. PMID 18767168. (Review).
- G. B. Irvine, O. M. El-Agnaf u.a.: Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. In: Molecular medicine (Cambridge, Mass.). Band 14, Nummer 7–8, 2008 Jul-Aug, S. 451–464, ISSN 1076-1551. doi:10.2119/2007-00100.Irvine. PMID 18368143. PMC 2274891. (Review).
- E. Laskowska, E. Matuszewska, D. Kuczynska-Wisnik: Small heat shock proteins and protein-misfolding diseases. In: Current pharmaceutical biotechnology. Band 11, Nummer 2, Februar 2010, S. 146–157, ISSN 1873-4316. PMID 20166966.
- N. Gregersen: Protein misfolding disorders: pathogenesis and intervention. In: Journal of inherited metabolic disease. Band 29, Nummer 2–3, 2006 Apr-Jun, S. 456–470, ISSN 1573-2665. doi:10.1007/s10545-006-0301-4. PMID 16763918. (Review).
- M. Stefani: Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. In: Biochimica et biophysica acta. Band 1739, Nummer 1, Dezember 2004, S. 5–25, ISSN 0006-3002. doi:10.1016/j.bbadis.2004.08.004. PMID 15607113. (Review).
Einzelnachweise
- ↑ D. M. Cyr, D. N. Hebert: Protein quality control–linking the unfolded protein response to disease. Conference on 'From Unfolded Proteins in the Endoplasmic Reticulum to Disease'. In: EMBO reports. Band 10, Nummer 11, November 2009, S. 1206–1210, ISSN 1469-3178. doi:10.1038/embor.2009.224. PMID 19851332. PMC 2775177.
- ↑ U. Schubert, L. C. Antón u.a.: Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. In: Nature. Band 404, Nummer 6779, April 2000, S. 770–774, ISSN 0028-0836. doi:10.1038/35008096. PMID 10783891.
- ↑ S. B. Qian, J. R. Bennink, J. W. Yewdell: Quantitating defective ribosome products. In: Methods in molecular biology (Clifton, N.J.). Band 301, 2005, S. 271–281, ISSN 1064-3745. doi:10.1385/1-59259-895-1:271. PMID 15917638.
- ↑ J. W. Yewdell, L. C. Antón, J. R. Bennink: Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? In: Journal of immunology. Band 157, Nummer 5, September 1996, S. 1823–1826, ISSN 0022-1767. PMID 8757297. (Review).
- ↑ K. F. Winklhofer, J. Tatzelt, C. Haass: The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. In: The EMBO journal. Band 27, Nummer 2, Januar 2008, S. 336–349, ISSN 1460-2075. doi:10.1038/sj.emboj.7601930. PMID 18216876. PMC 2234348. (Review).
- ↑ R. Kisilevsky: Amyloids: tombstones or triggers? In: Nature medicine. Band 6, Nummer 6, Juni 2000, S. 633–634, ISSN 1078-8956. doi:10.1038/76203. PMID 10835676. (Review).
- ↑ a b c Y. L. Lyubchenko, B. H. Kim u.a.: Nanoimaging for protein misfolding diseases. In: Wiley interdisciplinary reviews. Nanomedicine and nanobiotechnology. Band 2, Nummer 5, 2010 Sep-Oct, S. 526–543, ISSN 1939-0041. doi:10.1002/wnan.102. PMID 20665728. (Review).
- ↑ L. Schöls, O. Riess, T. Schmidt: Autosomal dominant vererbte spinozerebellare Ataxien: Klinik, Genetik und Pathogenese. In: Dtsch Arztebl. Band 98, Nummer 23, 2001, S. A-1546 / B-1319 / C-1233.
- ↑ V. Tarlac, E. Storey: Role of proteolysis in polyglutamine disorders. In: Journal of neuroscience research. Band 74, Nummer 3, November 2003, S. 406–416, ISSN 0360-4012. doi:10.1002/jnr.10746. PMID 14598317. (Review).
- ↑ M. Ramírez-Alvarado, J. N. Buxbaum: Systemic Amyloidose. In: M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9, S. 325-346. Eingeschränkte Vorschau in der Google Buchsuche
- ↑ V. L. Sim, B. Caughey: Prion Disease Therapy: Trials and Tribulations. In: M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9, S. 259-304. Eingeschränkte Vorschau in der Google Buchsuche
- ↑ J. Wang, J. A. King: Cataract as a Protein-Aggregation Disease. In: M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9, S. 487-516. Eingeschränkte Vorschau in der Google Buchsuche
- ↑ P. B. Stathopulos, J. A. Rumfeldt u.a.: Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. In: Proceedings of the National Academy of Sciences of the United States of America. Band 100, Nummer 12, Juni 2003, S. 7021–7026, ISSN 0027-8424. doi:10.1073/pnas.1237797100. PMID 12773627. PMC 165823.
- ↑ L. Wang, K. J. Colodner, M. B. Feany: Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. In: The Journal of neuroscience. Band 31, Nummer 8, Februar 2011, S. 2868–2877, ISSN 1529-2401. doi:10.1523/JNEUROSCI.3410-10.2011. PMID 21414908. PMC 3082397.
- ↑ D. Hervé, H. Chabriat: CADASIL. In: Journal of geriatric psychiatry and neurology. Band 23, Nummer 4, Dezember 2010, S. 269–276, ISSN 0891-9887. doi:10.1177/0891988710383570. PMID 21045164. (Review).
- ↑ A. A. Amato, R. J. Barohn: Inclusion body myositis: old and new concepts. In: Journal of neurology, neurosurgery, and psychiatry. Band 80, Nummer 11, November 2009, S. 1186–1193, ISSN 1468-330X. doi:10.1136/jnnp.2009.173823. PMID 19864656. (Review).
- ↑ L. Skora: High-resolution characterization of structural changes involved in prion diseases and dialysis-related amyloidosis. Dissertation, Georg-August-Universität Göttingen, 2009, S. iii.
- ↑ P. J. Waters: Degradation of mutant proteins, underlying "loss of function" phenotypes, plays a major role in genetic disease. In: Current issues in molecular biology. Band 3, Nummer 3, Juli 2001, S. 57–65, ISSN 1467-3037. PMID 11488412. (Review).
- ↑ a b D. N. Hebert, M. Molinari: In and out of the ER: protein folding, quality control, degradation, and related human diseases. In: Physiological reviews. Band 87, Nummer 4, Oktober 2007, S. 1377–1408, ISSN 0031-9333. doi:10.1152/physrev.00050.2006. PMID 17928587. (Review).
- ↑ R. R. Kopito: Biosynthesis and degradation of CFTR. In: Physiological reviews. Band 79, Nummer 1, 1999, S. S167–S173, ISSN 0031-9333. PMID 9922380. (Review).
- ↑ F. Melchior: Intrazellulärer Protein Abbau. Zentrum für Molekulare Biologie, WS 2009/2010, S. 30.
- ↑ L. Al-Bhalal, M. Akhtar: Molecular basis of autosomal dominant polycystic kidney disease. In: Advances in anatomic pathology. Band 12, Nummer 3, Mai 2005, S. 126–133, ISSN 1072-4109. PMID 15900113. (Review).
- ↑ H. M. McLaughlin, R. Sakaguchi u.a.: A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with charcot-marie-tooth disease type 2N (CMT2N). In: Human mutation. [elektronische Veröffentlichung vor dem Druck] Oktober 2011, ISSN 1098-1004. doi:10.1002/humu.21635. PMID 22009580.
- ↑ C. Li, C. Iosef u.a.: Disease-causing SAP mutants are defective in ligand binding and protein folding. In: Biochemistry. Band 42, Nummer 50, Dezember 2003, S. 14885–14892, ISSN 0006-2960. doi:10.1021/bi034798l. PMID 14674764.
- ↑ S. Kjaer, C. F. Ibáñez: Intrinsic susceptibility to misfolding of a hot-spot for Hirschsprung disease mutations in the ectodomain of RET. In: Human molecular genetics. Band 12, Nummer 17, September 2003, S. 2133–2144, ISSN 0964-6906. doi:10.1093/hmg/ddg227. PMID 12915470.
- ↑ J. P. Lerner-Ellis, J. C. Tirone u.a.: Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. In: Nature genetics. Band 38, Nummer 1, Januar 2006, S. 93–100, ISSN 1061-4036. doi:10.1038/ng1683. PMID 16311595.
- ↑ E. M. Bongers, P. H. Duijf u.a.: Mutations in the human TBX4 gene cause small patella syndrome. In: American journal of human genetics. Band 74, Nummer 6, Juni 2004, S. 1239–1248, ISSN 0002-9297. doi:10.1086/421331. PMID 15106123. PMC 1182087.
- ↑ E. Piters, C. Culha u.a.: First missense mutation in the SOST gene causing sclerosteosis by loss of sclerostin function. In: Human mutation. Band 31, Nummer 7, Juli 2010, S. E1526–E1543, ISSN 1098-1004. doi:10.1002/humu.21274. PMID 20583295.
- ↑ W. E. Balch, I. Braakman u.a.: Folding Biology of Cystic Fibrosis: A Consortium-Based Approach to Disease. In: M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9, S. 403-424. Eingeschränkte Vorschau in der Google Buchsuche
- ↑ D. M. Hutt, D. Herman u.a.: Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. In: Nature chemical biology. Band 6, Nummer 1, Januar 2010, S. 25–33, ISSN 1552-4469. doi:10.1038/nchembio.275. PMID 19966789. PMC 2901172.
- ↑ S. W. Gersting, K. F. Kemter u.a.: Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. In: American journal of human genetics. Band 83, Nummer 1, Juli 2008, S. 5–17, ISSN 1537-6605. doi:10.1016/j.ajhg.2008.05.013. PMID 18538294. PMC 2443833.
- ↑ B. Utsch, C. D. McCabe u.a.: Molecular characterization of HOXA13 polyalanine expansion proteins in hand-foot-genital syndrome. In: American journal of medical genetics. Band 143A, Nummer 24, Dezember 2007, S. 3161–3168, ISSN 1552-4833. doi:10.1002/ajmg.a.31967. PMID 17935235.
- ↑ F. Wang, W. Song u.a.: Inhibition of ER-associated degradation rescues native folding in loss of function protein misfolding diseases. In: The Journal of biological chemistry. [elektronische Veröffentlichung vor dem Druck] Oktober 2011, ISSN 1083-351X. doi:10.1074/jbc.M111.274332. PMID 22006919.
- ↑ T. Edmunds u.a.: Gaucher Disease. In: M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9, S. 403-424. Eingeschränkte Vorschau in der Google Buchsuche
- ↑ G. Hin-Fai Yam u.a.: Fabry disease: on the molecular pathogenesis of a lysosomal storage disease and the use of a chemical chaperone for therapeutic intervention. Universität Zürich, vom 2. April 2007
- ↑ F. Wang, W. Song u.a.: Inhibition of ER-associated degradation rescues native folding in loss of function protein misfolding diseases. In: The Journal of biological chemistry. [elektronische Veröffentlichung vor dem Druck] Oktober 2011, ISSN 1083-351X. doi:10.1074/jbc.M111.274332. PMID 22006919.
- ↑ D. A. Wwnger: Krabbe disease. In: R. A. Pagon, T. D. Bird u.a.: (Hrsg.): GeneReviews. PMID 20301416
- ↑ V. E. Walker, M. J. Wong u.a.: Hsp40 chaperones promote degradation of the HERG potassium channel. In: The Journal of biological chemistry. Band 285, Nummer 5, Januar 2010, S. 3319–3329, ISSN 1083-351X. doi:10.1074/jbc.M109.024000. PMID 19940115. PMC 2823420.
- ↑ R. S. Williams, D. I. Chasman u.a.: Detection of protein folding defects caused by BRCA1-BRCT truncation and missense mutations. In: The Journal of biological chemistry. Band 278, Nummer 52, Dezember 2003, S. 53007–53016, ISSN 0021-9258. doi:10.1074/jbc.M310182200. PMID 14534301.
- ↑ D. A. Lomas, D. H. Perlmutter: Alpha-1-Antitrypsin Deficiency. In: M. Ramírez-Alvarado, J. W. Kelly, C. M. Dobson (Hrsg.): Protein Misfolding Diseases. Verlag John Wiley and Sons, 2010, ISBN 0-471-79928-9, S. 403-424. Eingeschränkte Vorschau in der Google Buchsuche
- ↑ C. M. Greene, S. D. Miller u.a.: Alpha-1 antitrypsin deficiency: a conformational disease associated with lung and liver manifestations. In: Journal of inherited metabolic disease. Band 31, Nummer 1, Februar 2008, S. 21–34, ISSN 1573-2665. doi:10.1007/s10545-007-0748-y. PMID 18193338. (Review).
- ↑ F. E. Cohen, J. W. Kelly: Therapeutic approaches to protein-misfolding diseases. In: Nature. Band 426, Nummer 6968, Dezember 2003, S. 905–909, ISSN 1476-4687. doi:10.1038/nature02265. PMID 14685252. (Review).
- ↑ T. K. Chaudhuri, S. Paul: Protein-misfolding diseases and chaperone-based therapeutic approaches. In: The FEBS journal. Band 273, Nummer 7, April 2006, S. 1331–1349, ISSN 1742-464X. doi:10.1111/j.1742-4658.2006.05181.x. PMID 16689923. (Review).
- ↑ Klinische Studie (Phase III): Study of the Effects of Oral AT1001 (Migalastat Hydrochloride) in Patients With Fabry Disease bei Clinicaltrials.gov der NIH
- ↑ B. E. Roberts, J. Shorter: Escaping amyloid fate. In: Nature structural & molecular biology. Band 15, Nummer 6, Juni 2008, S. 544–546, ISSN 1545-9985. doi:10.1038/nsmb0608-544. PMID 18523464.
- ↑ D. E. Ehrnhoefer, M. Duennwald u.a.: Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models. In: Human molecular genetics. Band 15, Nummer 18, September 2006, S. 2743–2751, ISSN 0964-6906. doi:10.1093/hmg/ddl210. PMID 16893904.
- ↑ a b D. E. Ehrnhoefer, J. Bieschke u.a.: EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. In: Nature structural & molecular biology. Band 15, Nummer 6, Juni 2008, S. 558–566, ISSN 1545-9985. doi:10.1038/nsmb.1437. PMID 18511942.
- ↑ J. Bieschke, J. Russ u.a.: EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. In: Proceedings of the National Academy of Sciences of the United States of America. Band 107, Nummer 17, April 2010, S. 7710–7715, ISSN 1091-6490. doi:10.1073/pnas.0910723107. PMID 20385841. PMC 2867908.
- ↑ Substanz EGCG in grünem Tee verhindert tödliche Plaquebildung bei Parkinson und Alzheimer - Erste Ergebnisse im Reagenzglas. Pressemitteilung des Max-Delbrück-Centrum für Molekulare Medizin vom 30. Mai 2008
- ↑ K. Rezai-Zadeh, G. W. Arendash u.a.: Green tea epigallocatechin-3-gallate (EGCG) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. In: Brain research. Band 1214, Juni 2008, S. 177–187, ISSN 0006-8993. doi:10.1016/j.brainres.2008.02.107. PMID 18457818.
Weblinks
- L. Walker: Proteopathies: Protein Conformational Diseases. Vom 18. September 2008



Wikimedia Foundation.