- 2-FDG
-
Strukturformel 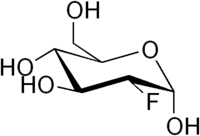
Allgemeines Freiname Fludeoxyglucose (18F) Andere Namen - Fluorodeoxyglucose
- Fluor-Desoxyglucose
- 18F-2-Fluor-2-deoxy-D-glucose
- 2-Deoxy-2-fluorglucose
- 2-Desoxyfluorglucose
- FDG
- 2-FDG
Summenformel C6H11FO5 CAS-Nummer - 29702-43-0
- 63503-12-8 (18F)
ATC-Code V09IX04
Eigenschaften Molare Masse 182,15 g·mol−1
181,15 g·mol−1 (18F)Aggregatzustand fest
Schmelzpunkt 170–176 °C [1]
Löslichkeit löslich in Wasser
Sicherheitshinweise Gefahrstoffkennzeichnung [1] 
Reizend (Xi) nur für 19F
R- und S-Sätze R: 36/37/38 für 19F S: 26-36 für 19F Bitte beachten Sie die eingeschränkte Gültigkeit der Gefahrstoffkennzeichnung bei Arzneimitteln Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Fluordesoxyglucose oder Fluor-Deoxyglucose oder Fluorodeoxyglucose ist ein Glucose-Analogon. Der vollständige Name der Verbindung lautet 2-Fluor-2-deoxy-D-glucose und wird üblicherweise mit FDG oder 2-FDG abgekürzt. Besondere Bedeutung hat das radioaktive 18F-2-FDG. Es ist das mit Abstand am häufigsten verwendete Radiopharmakon für die Erstellung einer Positronen-Emissions-Tomographie (PET). Es wird zur Messung des regionalen Glucoseverbrauchs mittels PET in der Kardiologie, der Neurologie und der Onkologie verwendet.[2]
Das natürliche Fluoratom mit der Nukleonenzahl 19 ist im FDG-Molekül in diesem Fall durch das radioaktive Fluor-18 Isotop ersetzt. Dieses Isotop ist ein Positronen-Emitter mit einer Halbwertszeit von lediglich 109,8 Minuten. Es ist in der Natur nicht vorhanden. Zur Herstellung von 18F-2-FDG wird dieses Isotop meist mit Hilfe eines Zyklotrons gewonnen, beispielsweise durch Beschuss des schweren Sauerstoff-Isotopes 18O mit Protonen.
Inhaltsverzeichnis
Metabolismus von FDG
18F-Fluor-Deoxyglukose wird von den Zellen des menschlichen Körpers wie Glukose aufgenommen, obwohl an einer Stelle des Moleküls eine Hydroxylgruppe durch das Radionuklid 18F ersetzt ist. Dabei wird FDG von den Zellen passiv mittels Glucosetransporter aus dem Blut aufgenommen. Derzeit sind beim Menschen fünf dieser Transporter bekannt. Sie werden mit Glut-1 bis Glut-5 bezeichnet.[3] Der Glut-1 ist das wichtigste Transportprotein für die Aufnahme von FDG in Tumoren und normalem Hirngewebe. Die Aufnahme in der Skelettmuskulatur und im Herzmuskel ist durch Insulin stimulierbar und erfolgt über den Glut-4 Transporter.[4] Das Enzym Hexokinase phosphoryliert FDG anschließend innerhalb der Zelle. FDG kann allerdings von den Zellen nach der Phosphorylierung nicht weiter verstoffwechselt werden. Die Rückreaktion, die Dephosphorylierung von FDG-6-Phosphat zu FDG, erfolgt – mit Ausnahme der Leber – in allen Organen und im Tumorgewebe sehr langsam.[4] Deshalb findet eine Anreicherung von FDG-6-Phosphat in den Zellen statt (metabolic trapping). Anhand des Zerfalls von 18F kann FDG detektiert werden. Die Verteilung von FDG im Körper erlaubt Rückschlüsse auf den Glukosestoffwechsel verschiedener Gewebe. Dies ist besonders für die frühe Diagnose von Krebserkrankungen von Vorteil, da eine Tumorzelle typischerweise aufgrund eines erhöhten Stoffwechsels viel Glukose verbraucht und dementsprechend FDG anreichert.
Als kleines Molekül überwindet FDG problemlos die Blut-Hirn-Schranke. Da das menschliche Gehirn einen hohen Bedarf an Glucose hat, wird ein entsprechend großer Anteil von FDG im Gehirn angereichert. Des weiteren reichert sich beim gesunden Menschen FDG in den Nieren und den ableitenden Harnwegen an.
Da 18F in 18O zerfällt, bildet sich aus FDG nach dem Zerfall und Aufnahme eines freien Wasserstoffatoms aus der Umgebung zur Bildung einer OH-Gruppe „normale“ Glucose mit einem schweren Sauerstoffkern, der mit 0,2 % Häufigkeit ein natürlich vorkommendes Isotop des Sauerstoffs ist. Die gebildete Glucose wird anschließend auf normalem Wege metabolisiert.
Die durch den Zerfall von 18F-FDG mögliche Bildgebung ist ein Abbild der Verteilung der Glucose-Aufnahme und -Phosphorylierung der Zellen im menschlichen Körper.
Synthese
Da eine direkte Bestrahlung normaler Glucose mit hochenergetischen Protonen zur Bildung von 18F-FDG lediglich zur Zerstörung des organischen Moleküls führt, muss das radioaktive Isotop getrennt mit Hilfe eines Zyklotrons hergestellt und anschließend weiter verarbeitet werden.
Die Synthese von 2-FDG ist wegen der Randbedingungen
- kurze Halbwertszeit des 18F,
- Strahlenexposition und
- Reinheit und Sterilität der Injektionslösung
sehr anspruchsvoll und aufwändig. Mittlerweile ist die Präparation von 2-FDG weitgehend automatisiert. Eine detaillierte Qualitätskontrolle, wie beispielsweise eine Keimzahluntersuchung ist, bedingt durch die kurze Halbwertszeit, kaum möglich.
Für die Herstellung von 18F-2-Fluor-2-deoxy-D-glucose sind eine Reihe von möglichen Reaktionen beschrieben. In der Praxis haben sich zwei Synthesewege bewährt: Die Elektrophile Addition, das heißt die Addition von 18F-F2 an Doppelbindungen und die Nukleophile Substitution mit 18F−. Einen guten Überblick über die Synthese und Qualitätskontrolle von 18F-FDG gibt ein Review-Artikel von S. Yu.[5] Der Weg über die Nukleophile Substitution sei hier exemplarisch beschrieben.
Herstellung von 18F−
In einem Zyklotron wird ein Target aus mit dem Sauerstoffisotop 18O angereichertem Wasser H218O mit hochenergetischen Protonen (ca. 15 MeV) bombardiert. Dabei wird in einer Kernreaktion ein kleiner Teil des 18O-Sauerstoffs unter Aufnahme je eines Protons und der Abgabe eines Neutrons in das radioaktive Fluorisotop 18F umgewandelt. Das 18F Isotop liegt in der wässrigen Phase als Fluorid-Ion 18F− vor.
Umsetzung mit einem 2-FDG-Precursor
Über einen Ionenaustauscher wird das gebildete Fluorid vom Wasser abgetrennt und anschließend mit einer Lösung aus Acetonitril, Kryptofix 222 und Kaliumcarbonat vom Ionenaustauscher wieder abgetrennt (eluiert).
In der eigentlichen nukleophilen Substitution ersetzt das radioaktive Fluorid-Ion eine leicht zu entfernende Abgangsgruppe, wie beispielsweise ein Triflat. Ein geeigneter Precursor für die Herstellung von 2-FDG mittels nukleophiler Substitution ist 1,3,4,6-O-Acetyl-2-O-trifluormethansulfonyl-β-D-mannopyranose, kurz als Mannose-Triflat bezeichnet. Nach der Substitution des Triflates werden die vier Acetyl-Schutzgruppen mittels basischer oder saurer Hydrolyse mit verdünnter Natronlauge bzw. verdünnter Salzsäure entfernt.
Aufreinigung
Durch die reverse phase-HPLC kann das gebildete 2-FDG sauber von den Ausgangsstoffen abgetrennt werden.
Anwendung
 Ganzkörper PET-Aufnahme mit 18F-Fluor-Desoxyglucose
Ganzkörper PET-Aufnahme mit 18F-Fluor-Desoxyglucose2-FDG wird in der PET für die Diagnose, Staging (Stadienbestimmung), Therapieeinstellung und Therapiekontrolle verwendet. Man spricht in diesem Zusammenhang auch oft von der „FDG-PET“.
Neben der Hauptanwendung in der Onkologie wird FDG auch für die Diagnose der Alzheimerschen Krankheit[6], der Parkinsonschen Krankheit[7], der Chorea Huntington[7], von Epilepsien[6], in der Kardiologie[6] und in der Entzündungsdiagnostik[6] verwendet, allerdings in weit geringerem Maße als in der Onkologie.
Geeignete und nicht-geeignete Fragestellungen
Für die FDG-PET gibt es drei Hauptindikationen für die Untersuchung von Patienten mit onkologischen Erkrankungen[4]:
- die Differenzierung benigner/maligner (gutartige/bösartig) Tumoren,
- das Tumorstaging (Lymphknoten und Fernmetastasen) und
- die Differenzierung Narbengewebe/vitales Tumorgewebe (Rezidiv, residueller Tumor).
Insbesondere sehr langsam wachsende Tumoren weisen in der Regel keine wesentlich erhöhte FDG-Aufnahme aus. Eine FDG-PET-Untersuchung ist daher meist nur in Ausnahmefällen sinnvoll bei[4]:
- Prostatakarzinomen,
- differenzierten neuroendokrinen Tumoren (z. B. Karzinoid),
- Bronchoalveolären Karzinome,
- niedrig malignen Non-Hodgkin-Lymphomen,
- niedrig malignen Hirntumoren (Astrozytom II, Oligodendrogliom II) und
- Hepatozellulären Karzinomen (vor allem höher differenzierte Formen).
Floride Entzündungen bzw. Heilungsprozesse zeigen neben dem Tumorgewebe ebenfalls eine erhöhte FDG-Aufnahme. Eine Untersuchung zur Differenzierung beispielsweise von Abszessen und Tumorgewebe, Sarkoidose, Bronchialkarzinomen usw., kann deshalb kaum sinnvoll durchgeführt werden.[4]
Wie bereits erwähnt, führen hohe Blutzuckerspiegel zu einer reduzierten Aufnahme von FDG im Tumorgewebe.[3] Bei Nüchternblutzuckerwerten über 150 mg/dl wird deshalb die Indikation zur FDG-PET meist kritisch überprüft.[4]
Nach Abschluss einer Chemo- oder Strahlentherapie kommt es auch bei vitalen Tumorzellen häufig zu einer Reduktion der FDG-Aufnahme. Deshalb sollte zwischen PET-Untersuchung und Abschluss der Therapie ein Zeitraum von mindestens vier Wochen liegen. Eine Ausnahme sind Verlaufsuntersuchungen bzw. bestimmte klinische Studien.[4]
Applikation
Im Fall des Ganzkörper-Scans auf der Suche nach Tumoren oder deren Metastasen wird eine Dosis von etwa 200 bis 400 MBq über eine isotonische Kochsalzlösung in eine Vene des Patienten injiziert. Über die Körperfläche des Patienten wird die zu applizierende Aktivitätsmenge errechnet. Die Zielgröße ist es dabei, etwa 210.000 Ereignisse pro Schicht im PET zu registrieren zu können.[4]
Der Patient muss vor der Applikation von FDG mindestens für sechs Stunden nüchtern bleiben, um einen möglichst niedrigen Blutzuckerspiegel zu haben. Diese Anforderung ist für einige Diabetiker problematisch, da die entsprechenden Kliniken meist keine PET-Untersuchung durchführen, wenn der Blutzuckerspiegel über 10 mmol/l liegt. Der venöse Blutzuckerspiegel wird vor jeder FDG-PET Untersuchung bestimmt.[4]
Nach erfolgter Injektion muss der Patient meist für eine Stunde in möglichst völliger Ruhe ohne körperliche Betätigung liegen, um die Verteilung von FDG im Körper zu gewährleisten. Muskuläre Anstrengungen würden FDG zu den entsprechenden Muskeln leiten und das Ergebnis verfälschen bzw. zu Artefakten bei der Bildgebung führen kann. Oft beobachtet man im Bereich der Zunge eine starke Anreicherung von FDG, was durch häufige und starke Schluckbewegungen der zum Teil unter extremen psychischen Stress stehenden Patienten bedingt ist.
Vor der Applikation können Wasser oder andere „kalorienfreie“ Getränke vom Patienten zu sich genommen werden. Unmittelbar vor Beginn der Aufnahme soll der Patient die Blase entleeren.[4]
Strahlenexposition
Die Strahlendosis bei einer PET mit 2-FDG beträgt ca. 7–10 mSv. Im Vergleich dazu beträgt die Strahlendosis bei einer kontrastverstärkten Computer-Tomographie ca. 20–40 mSv.[8] Die Höhe jener Dosis entspricht etwa der doppelten bis dreifachen Dosis der natürlichen Strahlenexposition, der die europäische Bevölkerung im Durchschnitt ausgesetzt ist (ca. 3 mSv pro Jahr). Das Risiko des Auftretens von Nebenwirkungen durch die Strahlung ist aus diesem Grunde vernachlässigbar klein.[9] Die effektive Dosis nach der intravenösen Injektion von FDG beträgt 2,0 x 10−2 mSv/MBq. Die höchste Strahlenexposition liegt dabei für die Harnblase bei 1,7 x 10−1 mSv/MBq.[10][11][12]
Nebenwirkungen
Es sind keine allergischen oder toxischen Nebenwirkungen bekannt.[8] Allein die extrem geringe Dosis an injiziertem 2-FDG, die im Bereich Picomol bis Nanomol liegt, schließt dies aus. Im Vergleich dazu bewegen sich die in der CT oder Magnetresonanztomographie verwendeten Mengen an Kontrastmitteln im Bereich von einigen Millimol, das heißt, sie liegen um 6 bis 9 Größenordnungen höher.
Geschichtliches
Tatsuo Ido vom Brookhaven National Laboratory beschrieb 1977 als Erster die Synthese von 18F-FDG.[13] Die Verbindung wurde von Abass Alavi im August 1976 an der University of Pennsylvania zwei Freiwilligen injiziert. Erst Aufnahmen des Gehirns wurden mit einem gewöhnlichen Nuklear-Scanner (nicht-PET-Scanner) durchgeführt und konnten die Konzentration von FDG in diesem Organ erstmals darstellen.
Differenzierung Diagnose – Therapie
FDG ist als Diagnostikum eine außerordentlich nützliche und vielfach bewährte Verbindung. Die Anwendung hat einen rein diagnostischen Hintergrund. Für die Therapie (Strahlentherapie, in diesem besonderen Fall würde man von einer Endoradiotherapie sprechen) ist 18F-FDG vom Strahlungstyp her völlig ungeeignet.
Einzelnachweise
- ↑ a b Sigma-Aldrich Co.
- ↑ Anlagen zum Positionspapier der Fachgruppe Nuklearchemie in der Gesellschaft Deutscher Chemiker
- ↑ a b G. Lienhard u.a., How Cells Absorb Glucose, in Scientific American, 1992, S. 34–39
- ↑ a b c d e f g h i j Positronen-Emissions-Tomographie (PET) bei onkologischen Fragestellungen
- ↑ Review of 18F-FDG synthesis and quality control
- ↑ a b c d Klinische Anwendung der PET – Onkologie, Neurologie, Kardiologie und Entzündungsdiagnostik
- ↑ a b Medizin-Netz.de
- ↑ a b Clinical application of FDG-PET Scan
- ↑ Wie hoch ist die Strahlenbelastung?
- ↑ Radiation Dose to Patients from Radiopharmaceuticals, ICRP Publication 53, Pergamon Press, 1987
- ↑ Recommendations of the International Commission on Radiological Protection, ICRP Publication 60, Pergamon Press, 1990
- ↑ L. Johansson u.a. Effective dose from radiopharmaceuticals", in Eur J Nucl Med, 19/1992, S. 933–938
- ↑ T. Ido et al., Fluorination with F2-2. A convenient synthesis of 2-FDG, in J Org Chem 42/1977, S. 2341–2342
Literatur
- K. Wienhard: PET Grundlagen und Anwendungen der Positronen-Emissions-Tomographie, 1989, Springer-Verlag, ISBN 3-540-19451-7 bzw. ISBN 0-387-19451-7
- The Conception of FDG-PET Imaging von Abass Alavi und Martin Reivich
- E. Bustamante u.a. High Aerobic Glycolysis of Rat Hepatoma Cells in Culture: Role of Mitochondrial Hexokinase in PNAS, 74/2005, S. 3735
Weblinks
- Universität Tübingen: Klinische Anwendung der PET (im Internet Archive; 2,38 MB)
- TU München: Fachinformationen über PET (im Internet Archive; 504 KB)

Wikimedia Foundation.