- Hochleistungsdünnschichtchromatographie
-
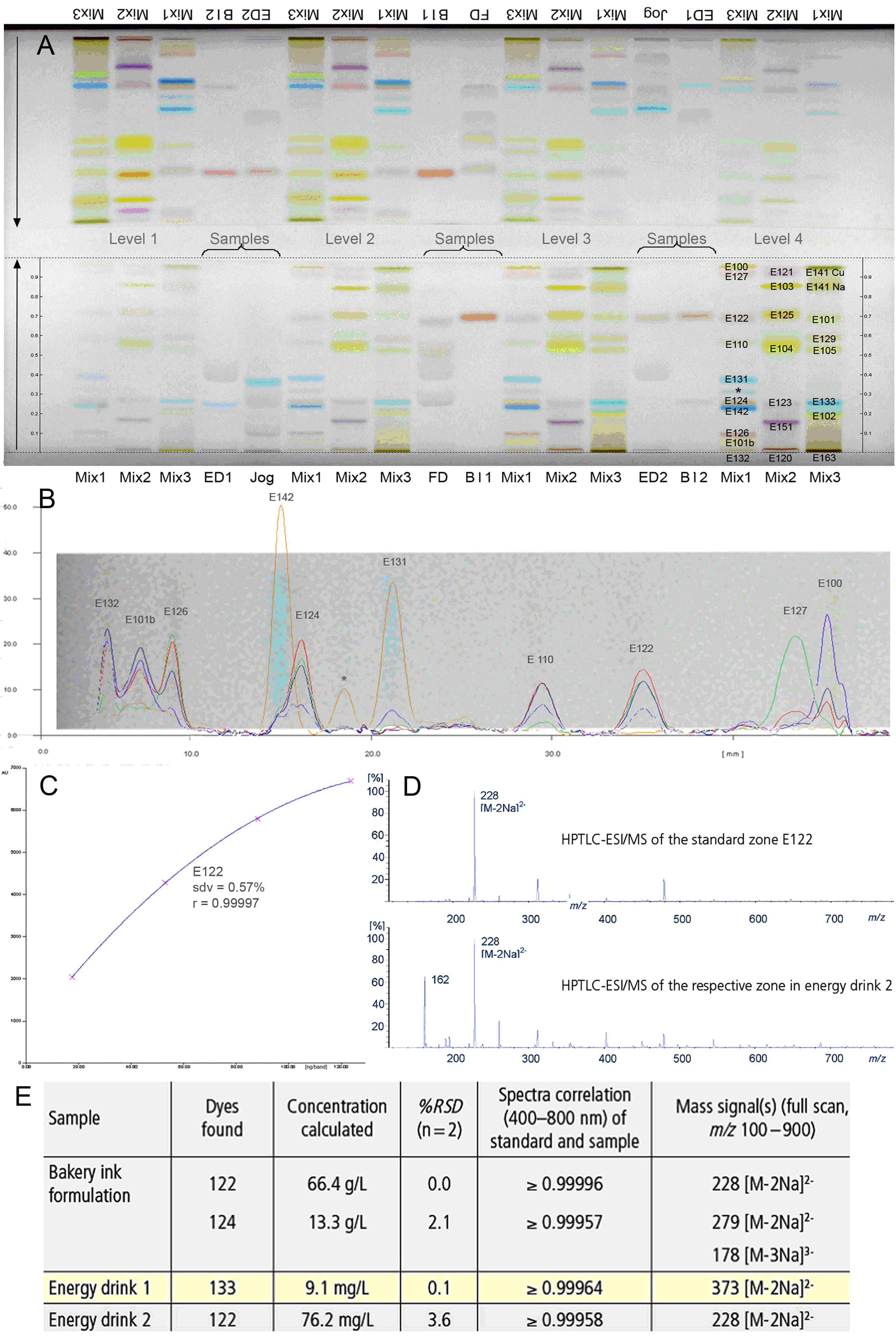
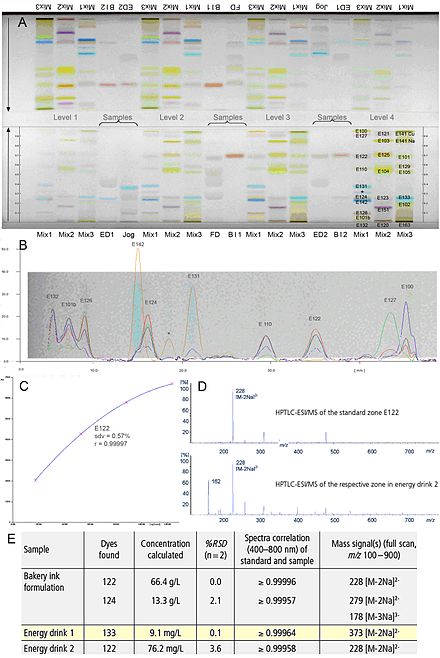 Abb. 1: Analytik von Lebensmittel-Farbstoffen in Proben: HPTLC-Chromatogramm von beiden Seiten entwickelt (A), Mehrwellenlängenscan von Mix 1 (B), Kalibrierfunktion (C), Massenspektren (D), quantitative Analyse (E)[1]
Abb. 1: Analytik von Lebensmittel-Farbstoffen in Proben: HPTLC-Chromatogramm von beiden Seiten entwickelt (A), Mehrwellenlängenscan von Mix 1 (B), Kalibrierfunktion (C), Massenspektren (D), quantitative Analyse (E)[1]
Die Hochleistungsdünnschichtchromatographie (HPTLC von engl. high-performance thin-layer chromatography) ist ein physikalisch-chemisches Trennverfahren und eine Weiterentwicklung der klassischen Dünnschichtchromatographie (DC), bei dem Hochleistungstrennschichten unter Einsatz von Geräten verwendet werden. Unter den planar-chromatographischen Methoden, zu denen noch die Papierchromatographie und die DC zählen, ist die HPTLC derzeit die leistungsstärkste.
Inhaltsverzeichnis
Geschichte
Bereits Mitte der 1960er Jahre gelang es, die DC als quantitative Methode zu verwenden.[2]
1975 wurde der Begriff HPTLC eingeführt,[3] als die ersten Fertigschichten eingeführt wurden. Der Begriff „HPTLC“ wird seitdem mit einer sehr hohen Trenneffizienz (Trennzahl max. 40), Präzision (typischerweise ≤ 2 %) und Detektierbarkeit (bis in den Pikogramm pro Zone-Bereich) verbunden. Der Begriff wird aber unterschiedlich gehandhabt; eine einheitliche Verwendung ist international noch nicht erkennbar. Teilweise wird die Auffassung vertreten, um HPTLC handele es sich nur, wenn ein apparativer Probenauftrag und eine Auswertung bei einer Trennung auf entsprechenden Platten vorgenommen wird, teilweise wird von HPTLC bei Verwendung entsprechender Trennschichten gesprochen.
1978 kamen modifizierte HPTLC-Fertigschichten und 1984 mit der automatisierten Mehrfachentwicklung eine trennleistungsstarke Entwicklungstechniken auf den Markt.
Sphärische HPTLC-Fertigschichten kamen 1995 hinzu, während monolithische Fertigschichten seit 2001 kommerziell erhältlich sind. Techniken mit Letzteren werden auch als „ultrathin-layer chromatography“ (UTLC) bezeichnet, da Merck diese Schichten unter diesem Handelsnamen vertreibt. UTLC bezeichnet aber auch die miniaturisierte Weiterentwicklung der Methode. Seit etwa 2000 wird auch an der Kopplung an die Massenspektrometrie gearbeitet.[4]
Die instrumentelle Entwicklung ist detailliert in einer chronologischen Listung erfasst.[5] Inzwischen sind alle HPTLC-Schritte (Auftragung, Entwicklung, Derivatisierung, Dokumentation, Densitometrie) standardisiert und automatisiert.
Grundlagen der Methode
Um die volle Leistungsstärke der HPTLC zu erreichen, sollten sowohl entsprechende Geräte zur Probenauftragung und Auswertung als auch HPTLC-Trennschichten in Kombination verwendet werden. Durch den Einsatz von, im Vergleich zur DC, leistungsstärkerem Trennmaterial (kleinere Korngröße von 5 bis 7 µm, engere Korngrößenverteilung, homogenere Schichtdicke), automatisierten Geräten für die einzelnen Arbeitsschritte und standardisierten Methoden ist es mit der HPTLC nicht nur möglich, eine qualitative, sondern auch eine schnelle quantitative Analyse von Proben aller Art durchzuführen (Abb. 1). Bei hohem Probendurchsatz ist z. B. die Trennzeit pro Probe 20 s bei einem Fließmittelverbrauch von 200 µl.[1]
HPTLC-Fertigschichten
 Abb. 2: Vielfalt der HPTLC-Fertigschichten
Abb. 2: Vielfalt der HPTLC-FertigschichtenDie stationäre Phase ist auf Trägermaterialien wie Glas oder Aluminiumfolien aufgebracht. Standardformate sind 10 cm × 10 cm bzw. 20 cm × 10 cm.
Die gebräuchlichste HPTLC-Fertigschicht ist Kieselgel, da sie für ca. 90 % aller HPTLC-Trennungen eingesetzt wird. Diverse Hersteller bieten verschiedene polare Kieselgelphasen an, z. B. wasserstabile, säurestabile oder hochreine Schichten. Die typische Schichtdicke ist 200 µm, es gibt aber auch dünnere Schichten von nur 100 oder 50 µm. Typischerweise verbessern sich auf dünneren Schichten die Detektierbarkeit und Laufzeit.
Die restlichen 10 % der HPTLC-Trennungen finden v. a. auf mittelpolaren und unpolaren Umkehrphasen (reversed phase, RP) RP-2-, 8- oder 18-Phasen statt (Abb. 2). Eine Besonderheit der mittelpolaren Schichten ist, dass diese wie Kieselgel für eine Normalphasentrennung eingesetzt werden können, aber in Kombination mit einem polareren Fließmittel auch für die Umkehrphasentrennung.
Beim Einsatz der HPTLC in der Spurenanalytik ist das Vorwaschen der Fertigschichten von Vorteil. Hierzu werden die Schichten in einem elutionsstarken Lösungsmittel chromatographiert, getrocknet, mit einer Gegenglasplatte bedeckt, in Aluminiumfolie gewickelt und bis zum Gebrauch in einem sauberen Exsikkator vor Kontaminationen geschützt gelagert.
Einfache Anpassung der Selektivität
Trennungen werden in der DC am effektivsten durch die Anpassung der Selektivität optimiert. Die Selektivitätsbreite ist in der DC/HPTLC einzigartig.
Man kann Fertigschichten in geeignete Imprägnierlösungen tauchen oder aber die mobile Phase der geforderten Selektivität anpassen. Die einsetzbaren Lösungsmittel, organische wie anorganische, sind vielfältig, und es ist keine Limitierung bezüglich der z. B. UV-Durchlässigkeit, Wassermischbarkeit, Viskosität oder MS-Gängigkeit gegeben. Effektive Selektivitätsanpassungen sind beispielsweise die:
- Trennung von Verbindungen nach v.a. Anzahl und Lage ihrer Doppelbindungen (Komplexstabilität) auf einer mit Silbernitrat imprägnierten Schicht.
- Trennung von polyaromatischen Verbindungen nach Charge-Transfer-Komplexstabilität auf einer mit Coffein imprägnierten Schicht.
- Trennung von Phenolen oder Säuren – zur schärferen Spotform – auf einer mit anorganischen Säuren imprägnierten Schicht oder von Alkaloiden oder Aminen auf einer mit anorganischen Basen imprägnierten Schicht.
Auftragung
Eine automatisierte, standardisierte, präzise und verschleppungsfreie Auftragung ist Voraussetzung für alle quantitativen HPTLC-Methoden. Das Auftragen der flüssig aufgearbeiteten Analysenprobe erfolgt am effektivsten automatisch, typischerweise durch bandförmiges Aufsprühen auf die HPTLC-Platte. Die Probe wird dabei mit Druckluft oder Stickstoff zerstäubt und der Einfluss des verwendeten Lösungsmittels durch das schnelle Abdampfen minimiert. Gleichzeitig verbessert sich durch die scharfe, bandenförmige Startzone – im Gegensatz zur punktförmigen Applikation – die Auflösung zwischen den einzelnen Komponenten.
Das Auftragevolumen der Probe kann von 100 nl bis 1 ml variieren. Gegenüber der DC sind die resultierenden Auftragemengen pro Zone deutlich reduziert, was die Auflösung verbessert. Nach der Auftragung erfolgt die homogene Trocknung der Startzonen. Voraussetzung für das Auftragen relativ großvolumiger, insbesondere wässriger oder matrixreicher, Analysenproben ist eine flächenförmige Auftragung. Zusätzlich kann die Auftragung bis 60 °C beheizt erfolgen. Flächenauftragung und beheiztes Auftragen reduzieren die erforderliche Auftragezeit signifikant.
Je nach Fließmittel ist dann vor der Trennung eine Frontelution der Analyten auf die Startzonen-Oberkante, sogenannte Fokussierung der Startzone, erforderlich (Dauer: einige Sekunden).
Eine sorgfältige Aufreinigung der Analysenprobe entsprechend der Säulenchromatographie HPLC (GC) ist nicht notwendig, denn Matrix (nicht interessierende Komponenten einer Probe) kann am Start verbleiben oder mit der Front wandern. Das einmalige Benutzen der Schicht ermöglicht die Beladung mit Matrix und so können Probenvorbereitung und Chromatographie sogar zeitgleich erfolgen.[6]
Entwicklung
Die Entwicklung der Platte mit dem Fließmittel, der eigentliche Trennvorgang, ist für das Ergebnis entscheidend. Die Standardisierung dieses Schrittes war wesentlich für die Reproduzierbarkeit der Methode, vor allem bei Luftfeuchte-anfälligen Systemen, z.B. wenn Kieselgelschichten mit unpolaren Fliessmitteln entwickelt werden. Moderne automatisierte Trennkammern kontrollieren die Plattenaktivität und das Kammerklima inklusive einer Vorkonditionierung der Platte, so dass reproduzierbare Chromatogramme routinemäßig erhalten werden. Durch Kapillarkräfte bewegt sich das Fließmittel durch die HPTLC-Schicht, nimmt lösliche Komponenten an den Startzonen auf und trennt diese je nach deren Wechselwirkungen mit dem Fließmittel und der stationären Phase im Verlauf der Entwicklung. Dabei erfolgt ein Abdampfen flüchtiger Fließmittel-Komponenten in den Dampfraum und ein Aufdampfen derselben auf den noch trockenen Teil der Schicht. Auch polare Komponenten des Fließmittels verarmen in einer Fließmittelmischung stärker als unpolare durch bevorzugte Adsorption an die aktive Schicht. Infolgedessen erfolgt bei Fließmittelgemischen eine unbewusste Gradiententrennung. Diese verläuft heute mit modernen automatischen Trennkammern reproduzierbar. Zudem wird die Laufstrecke automatisch überwacht und bei Erreichen der Endhöhe die Platte sofort und sehr homogen getrocknet. Laufstrecken (Abstand Startzonenmitte zu Frontlinie) sollten 60 mm nicht überschreiten, denn höhere Laufstrecken führen zwar zu einer besseren Zonentrennung, aber eben auch – durch Diffusion – zu einer größeren Zonenbreite und letztlich nicht zu einer verbesserten Auflösung zwischen Zonen. Zudem nimmt die Chromatographiezeit mit der Trennstrecke exponentiell zu, und der höhere Zeitaufwand rechtfertigt keinesfalls die höhere Trennstrecke. Weitaus sinnvoller ist es, die Selektivität einer Trennung zu optimieren und kurze Trennstrecken zu wählen. Hinsichtlich der Matrix ist eine Trennung so zu optimieren, dass Matrixkomponenten nicht mit dem Fließmittel wechselwirken und entweder an der Startzone zurückbleiben oder mit der Front wandern. Alle Probebestandteile (ausgenommen leichtflüchtige) sind auf Grund der offenen planaren Schicht der Detektion zugänglich. Bei nachträglichem Interesse z.B. an den Matrixzonen kann die gleiche Platte nochmals mit einem elutionsstärkeren Fließmittel chromatographiert werden oder bei Frontelution mit einem elutionsschwächeren. Diese Flexibilität in der Chromatographie, die umfassende Detektierbarkeit ansonsten unsichtbarer Probebestandteile (im Gegensatz zur HPLC) und die Analyse der weitgehend unveränderten Probe (reduzierte Probenvorbereitung) sind Stärken der HPTLC.
 Abb. 3: Multiple Detektion in der EU zugelassener Süßstoffe auf der gleichen HPTLC-Platte (Reagenzfolge): Quantifizierung (1) im UV 254 nm (Saccharin), (2) im UV 366 nm nach Derivatisierung mit Primulin-Reagenz (Acesulfam-K, Na-Cyclamat), im sichtbaren Bereich nach Derivatisierung mit (3) Ninhydrin-Reagenz (Aspartam) und (4) ß-Naphthol-Reagenz (Sucralose, Neohesperidin-Dihydrochalkon, Stevia (Rebaudiosid A))[7]
Abb. 3: Multiple Detektion in der EU zugelassener Süßstoffe auf der gleichen HPTLC-Platte (Reagenzfolge): Quantifizierung (1) im UV 254 nm (Saccharin), (2) im UV 366 nm nach Derivatisierung mit Primulin-Reagenz (Acesulfam-K, Na-Cyclamat), im sichtbaren Bereich nach Derivatisierung mit (3) Ninhydrin-Reagenz (Aspartam) und (4) ß-Naphthol-Reagenz (Sucralose, Neohesperidin-Dihydrochalkon, Stevia (Rebaudiosid A))[7]Dokumentation
Die Lage der getrennten Zonen wird anhand ihres Weißlichtbeleuchtung durch elektronische Dokumentationssysteme belegt.
Gegenüber der DC ist in der HPTLC die Substanzmenge pro Zone deutlich reduziert. So sind sichtbare Zonen bei visueller Betrachtung der Platte kaum sichtbar. Durch die elektronische Dokumentation werden auch schwache Zonen auf der Platte gut sichtbar, z.B. durch die Durchlichtaufnahme und Bildbearbeitungswerkzeuge.
Derivatisierung
Die problemlose Zugänglichkeit aller Proben und ihrer Komponenten zur prä- oder postchromatographischen Derivatisierung ist ein nicht zu unterschätzender Vorteil der DC/HPTLC. Zur weiteren Detektion nicht UV-aktiver (sichtbar durch Fluoreszenzindikatoren in der Schicht), nicht sichtbarer und nicht nativ fluoreszierender Substanzen, kann eine mikrochemische Derivatisierung durchgeführt werden. Diese erfolgt meist postchromatographisch, kann aber auch in situ prächromatographisch (direkt in der Startzone) erfolgen. In der HPTLC muss die Aufbringung des Derivatisierungsreagenzes homogen erfolgen, z.B. durch Tauchen oder Bedampfen, ebenso wie das bei Bedarf anschließende Erhitzen der Platte. Reagenzfolgen, d.h. die Detektion der gleichen Platte nacheinander mit unterschiedlichen Derivatisierungsreagenzien, sind möglich (Abb. 3) und zeigen die enorme Flexibilität der HPTLC hinsichtlich der Detektion. Weiterhin können biologische oder biochemische Derivatisierungen direkt auf der HPTLC-Platte durchgeführt werden.
Densitometrie (Densitogramm)
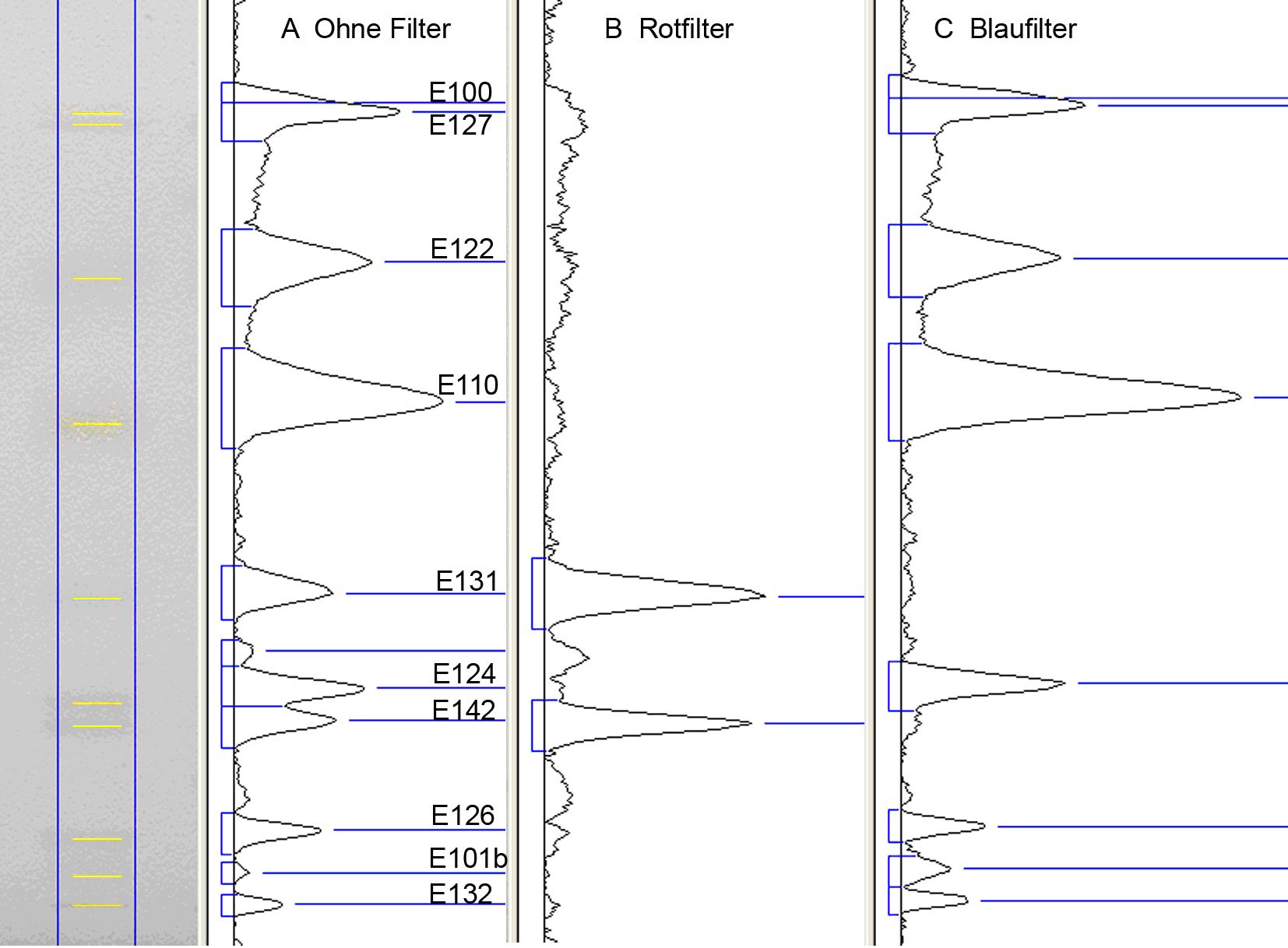
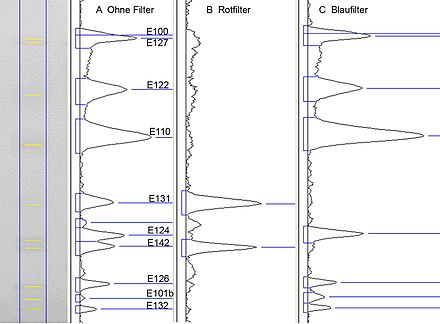 Abb. 4: Elektronische Bildauswertung einer dokumentierten Bahn mit wasserlöslichen Lebensmittel-Farbstoffen unter Einsatz von selektiven elektronischen Filtern[1]
Abb. 4: Elektronische Bildauswertung einer dokumentierten Bahn mit wasserlöslichen Lebensmittel-Farbstoffen unter Einsatz von selektiven elektronischen Filtern[1]Bei der klassischen Densitometrie wird das Chromatogramm von einem Scanner mit monochromatischem Licht bahnweise abgetastet. Das von der Oberfläche diffus gestreute Licht (Remission) wird von einem Photomultiplier detektiert. Bei der Absorptionsmessung absorbieren Substanzen in den Trennzonen Licht, und es kommt – im Vergleich zum Plattenhintergrund – weniger Licht am Detektor an. Dieses Signal wird invertiert (indirekte Messung). Demgegenüber wird bei der Fluoreszenzmessung ein direktes Signal erhalten. Das remittierte monochromatische Licht der Anregungswellenlänge wird vor dem Detektor ausgeblendet, und nur das von der Substanz ausgestrahlte Licht (Emissionswellenlänge) wird erfasst. Eine quantitative Auswertung wird mit Bezug auf Vergleichsstandards durchgeführt (Relativmessung). Kalibrierfunktionen sind bei Absorptionsmessungen überwiegend polynom – Gültigkeit hat hier in guter Näherung die Kubelka-Munk-Funktion[8] – bei Fluoreszenzmessungen sind die Kalibrierfunktionen meist linear. Eine besondere Stärke der klassischen Densitometrie liegt in ihrer spektralen Selektivität. So können sowohl Absorptionsspektren aufgenommen als auch ein Chromatogramm sequentiell mit verschiedenen Wellenlängen im Absorptions- wie Fluoreszenzmodus ausgewertet werden (Mehrwellenlängenscan). Es sein darauf hingewiesen, dass die höchste quantitative Präzision erzielt wird, wenn die Vermessung im Absorptionsmaximum der zu bestimmenden Substanz erfolgt. Die häufig angetroffene Absorptionsmessung bei 254 nm (Absorptionsmaximum der Fluoreszenzindikators in der Schicht) ist nicht sinnvoll, weil der Detektor (Photomultiplier) das remittierte UV erfasst und nicht – wie das menschliche Auge – das remittierte sichtbare Licht.
Die Auswertung mittels elektronischer Bilderfassung ist eine jüngere, sehr schnelle Variante der Erfassung des gesamten Trennbildes. Sie erfolgt im polychromatischen sichtbaren (weißen) Licht. Wird zur Beleuchtung langwelliges UV-Licht (z. B. UV 366 nm) verwendet, erfasst die Kamera die Fluoreszenz der Zonen im sichtbaren Bereich, wird kurzwelliges UV-Licht (z. B. UV 254 nm) für Schichten mit Fluoreszenzindikator verwendet, erfasst die Kamera – genau wie das menschliche Auge – die Fluoreszenzminderung des Fluoreszenzindikators durch Trennzonen, die in einer großen Bandweite um UV 254 nm absorbieren. Aus den Bilddaten ist eine Quantifizierung möglich. Dazu wird ein Bahnraster über das Bild gelegt und das Bild in Grauwerte umgerechnet. Über jede Zeile einer Bahn werden die Grauwerte summiert. Dadurch ergibt sich die Analogkurve. Die spektrale Selektivität ist auf die visuelle Farberkennung beschränkt, ermöglicht aber den Einsatz von selektiven Filtern (Abb. 4).
Vorteile und Grenzen
HPTLC ermöglicht eine effektive, kostengünstige und schnelle Analytik. Bereits angesprochen wurden folgende Vorteile:
- Probenvorbereitung während der Chromatographie
- Konzentrieren der Proben beim Auftragen (Aufsprühen hoher Volumina)
- Reduzierte Probenvorbereitung (durch die einmalige Verwendung der Schicht) ermöglicht die Analytik einer weitgehend unveränderten Probe
- Multiple Detektion
- Parallele Chromatographie unter identischen Bedingungen
Hinzu kommen Vorteile des flexiblen modularen offline-Prinzips:
- Der Probendurchsatz kann bei Bedarf auf 1000 Läufe pro 8 h-Tag ausgedehnt werden, indem im 20 min-Takt zwischen den automatisierten Arbeitsschritten gewechselt wird.
- Kopplungen (Hyphenations) sind leicht durchführbar, da das Fließmittel nicht stört und die Substanzen stationär gespeichert vorliegen, v.a. in Kombination mit Bioassays für die wirkungsorientierte Analytik.[9]
- Nach der Auswertung kann von ausgewählten Zonen das Massenspektrum aufgenommen werden. Es muss nicht a priori jeder Lauf, samt Matrix und Hintergrund, vermessen werden (status quo der Säulenchromatographie).
Nachteil der HPTLC ist die im Vergleich zu HPLC oder GC geringere Trennleistung. Jedoch ermöglicht die selektive Derivatisierung post-chromatographisch einen Gewinn an Trennleistung, indem man, um die Analyten zu detektieren, die „selektive Brille“ aufsetzt (z. B. derivatisiert oder elektronische Filter benutzt). Heute kann man über die Reinheit der Massenspektren oder UV/Vis-Spektren (Korrelation der an unterschiedlichen Stellen innerhalb eines Peaks gemessenen Spektren) prüfen, ob die Trennleistung ausreicht. In diversen analytischen Fragestellungen ist eine HPTLC-Methode von Vorteil.
Einzelnachweise
- ↑ a b c G. Morlock, C. Oellig, CAMAG Bibliography Service 103 (2009) 5.
- ↑ H. Jork, Direkte spektralphotometrische Auswertung von Dünnschicht-Chromatogrammen im UV-Bereich, Fresenius' Zeitschrift für Analytische Chemie 221 (1966) 17. doi:10.1007/BF00519562
- ↑ A. Zlatkis and R. E. Kaiser (Hrsg.), Journal of Chromatography Library, Vol. 9: HPTLC: High Performance Thin-Layer Chromatography, Elsevier Scientific Publishing Company, Amsterdam, 1977, S. 11.
- ↑ G.E. Morlock, W. Schwack, Coupling of planar chromatography to mass spectrometry, Trends Anal Chem 29_10 (2010) 1157-1171. doi:10.1016/j.trac.2010.07.010
- ↑ J. Sherma, G. Morlock, Chronology of thin-layer chromatography focusing on instrumental progress, J Planar Chromatogr 21 (2008) 471. doi:10.1556/JPC.21.2008.6.15
- ↑ G. Morlock, M. Vega: Two new derivatization reagents for planar chromatographic quantification of sucralose in dietetic products, J Planar Chromatogr 20 (2007) 411. doi:10.1556/JPC.20.2007.6.4
- ↑ G. Morlock, G. Shabier, J Chromatogr A (2010) invited.
- ↑ P. Kubelka, F. Munk, Z Techn Phys 12 (1931) 593.
- ↑ G. Morlock, W. Schwack: Hyphenations in planar chromatography. J Chromatogr A 1217 (2010) 6600-6609. doi:10.1016/j.chroma.2010.04.058.


Wikimedia Foundation.