- Massenspektrometrie
-
Als Massenspektrometrie werden Verfahren zum Messen der Masse von Atomen oder Molekülen bezeichnet. Die Massenspektrometrie zählt nicht zu den Methoden der Spektroskopie, da es nicht um Spektren von elektromagnetischer Strahlung geht.
Die zu untersuchende Substanz, der Analyt, wird in die Gasphase überführt und ionisiert. Die Ionen werden durch ein elektrisches Feld beschleunigt und dem Analysator zugeführt, der sie nach dem Masse-zu-Ladung-Verhältnis m/q "sortiert", beispielsweise (im Sektorfeld-Massenspektrometer, siehe unten) räumlich in Teilstrahlen auftrennt. Die Moleküle können dabei fragmentiert werden.[1][2]
Massenspektrometrie findet in vielen Bereichen Anwendung. Eingesetzt wird sie unter anderem bei der Charakterisierung von chemischen Verbindungen, in der medizinischen Chemie zur Identifizierung von Substanzen in Körperflüssigkeiten oder Organen, in kriminaltechnischen Untersuchungen, Dopingkontrollen, der militärischen Analytik von chemischen Kampfstoffen und in der Pharmakokinetik. Es gibt sehr unterschiedliche Techniken, die sich je nach Aufwand, Anwendung und Genauigkeit unterscheiden.
Vorteilhaft ist in vielen Bereichen, dass die Datenmenge recht gering ist und damit eine Kopplung mit Datenbanken von Massenspektren leicht möglich ist. Es ist auch verhältnismäßig leicht ein Massenspektrometer mit einer HPLC-Anlage oder einem Gaschromatographen zu koppeln und so die verschiedenen Massenspektren der im Analyten enthaltenden Verbindungen zu erhalten.
Geschichte
 Nachbau des dritten Massenspektrometers von J. J. Thomson
Nachbau des dritten Massenspektrometers von J. J. Thomson
Die Massenspektrometrie basiert auf einer Hypothese, die vom britischen Chemiker William Prout im frühen 19. Jahrhundert aufgestellt wurde und besagt, dass es eine Eigenschaft eines Atoms ist, eine bestimmte Masse zu haben – das Atomgewicht. Er hatte beobachtet, dass sich Atome von ihrer Masse her als ganzzahliges Vielfaches der Masse von Wasserstoff-Atomen verhalten. [3] [4] Spätere und genauere Messungen von Jöns Jakob Berzelius (1828) und Edward Turner (1832) schienen jedoch diese Hypothese zu widerlegen, denn es wurde z. B. für das Chlor-Atom eine Masse bestimmt, die das 35,45-fache der Wasserstoffmasse beträgt.
 Sir Joseph John Thomson
Sir Joseph John ThomsonIn der Mitte des 19. Jahrhunderts beobachtete Julius Plücker den Einfluss von magnetischen Feldern auf das Leuchten von Gasentladungsröhren.
Eugen Goldstein und Wilhelm Wien publizierten in den Jahren 1886 und 1898 die sogenannten Kanalstrahlen und ihre Ablenkung durch Felder. Sie hatten die weitreichenden Konsequenzen ihrer Entdeckungen jedoch 1886 noch nicht erkannt. [5] [6] [7] [8] [9] [10]
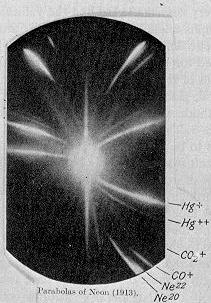
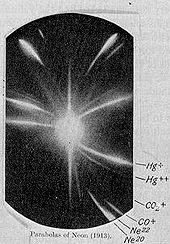 Frühe Fotoplatte mit den Isotopen von Neon (20Ne und 22Ne)
Frühe Fotoplatte mit den Isotopen von Neon (20Ne und 22Ne)Später, im Jahr 1897, publizierte J. J. Thomson verschiedene Experimente, in denen er in Vakuumröhren Kathodenstrahlen von verschiedenen Kathodenmetallen mit elektromagnetischen Feldern ablenkte, und stellte korrekte Gleichungen zum Zusammenhang zwischen Masse, Geschwindigkeit und Bahnradius auf. 1913 publizierte er eine Methode, um mit Hilfe eines Massenspektroskops Fotoplatten zu belichten und so qualitative und quantitative Untersuchungen an den in einer Röhre enthaltenen Gasen durchzuführen.
Ein Schüler von Thomson, der britische Chemiker und Physiker Francis William Aston, baute 1919 das erste funktionierende Massenspektrometer. Mit dieser neuen Technik konnte er die Isotope von Chlor (35Cl und 37Cl), von Brom (79Br und 81Br) und von Krypton (78Kr, 80Kr, 82Kr, 83Kr,84Kr und 86Kr) beobachten. Aston wurden schließlich im Jahr 1922 mit dem Nobelpreis für Chemie für seine Untersuchungen der Isotope geehrt. Durch die Verwendung der Technik des Elektrofokusierens konnte er nicht weniger als 212 der damals bekannten 287 Isotope beobachten.
Im Jahr 1918 wurde von Arthur Jeffrey Dempster das erste moderne Massenspektrometer entworfen und gebaut, welches 100 fach genauer arbeitete als alle vorherigen Entwicklungen und legte den Grundstein für das Design heutiger Massenspektrometer. Aufgrund dieser Entwicklung hat er im Jahr 1935 das das Uranisotop mit der Masse 235 identifizieren können.
Während des Manhattan-Projekts wurden riesige Isotopenanreicherungsanlagen auf Grundlage der Massenspektrometrie gebaut.
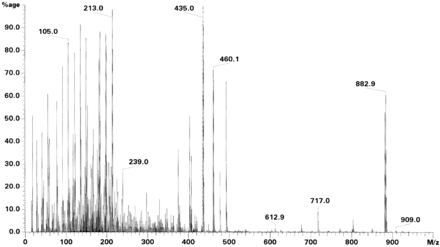 Stark Fragmentiertes Massenspektrum - Elektronenstoss-Ionisation
Stark Fragmentiertes Massenspektrum - Elektronenstoss-IonisationIn den 1950er Jahren wurde von Roland Gohlke und Fred McLafferty das erste Mal ein Massenspektrometer als Detektor für eine Chromatographie-Methode eingesetzt. Beide koppelten einen Gaschromatographen mit einem Massenspektrometer. Durch diese Methode konnten das erste Mal Substanzgemische in einer Anlage getrennt und identifiziert werden.[11][12] Für die Anwendung in einer gaschromatographischen Methode müssen die entsprechenden Verbindungen jedoch im Vakuum flüchtig und dabei unzersetzlich verdampfbar sein.
Im Jahr 1974 entwickelten Alan G. Marshall und Melvin B. Comisarow von der University of British Columbia inspiriert von den Fourier Transform Nuclear Magnetic Resonance (FT-NMR) und ICR Methoden ein Fouriertransform-Massenspektrometer.[13]
Die bisherigen Methoden zur Erzeugung der benötigten Ionen waren sehr aggressiv und führten zu vielen Bruchstücken (Fragmente) beim vermessen organischer Verbindungen. Daher setzte ab den 1960 Jahren die Entwicklung immer schonenderer Ionisationsmethoden ein. Mitte der 1960er Jahre wurde von Munson und Field die Chemische Ionisation (CI) veröffentlicht.[14] Im Jahr 1969 hat Beckey die Feld Desorbtion (FD) publiziert.[15]
Später erfolgten dann die weitere Entwicklung einer Vielzahl an Ionisationsmethoden für die unterschiedlichsten Zwecke wie zum Beispiel Chemische Ionisation bei Atmosphärendruck (APCI), Matrix-unterstützte Laser-Desorption/Ionisation (MALDI), Elektrospray (ESI).
Parameter eines Massenspektrometers
Ein Massenspektrometer wird im Wesentlichen durch zwei Parameter charakterisiert: Die Massenauflösung und die Massengenauigkeit.
Die Massenauflösung bezeichnet den minimalen Massenunterschied dm, den zwei Ionen haben müssen, damit sie noch aufgelöst werden können. Die Auflösung eines Massenspektrometers wird in der Einheit Thomson (Th) angegeben, wobei aber trotzdem öfter nur das Auflösungsvermögen R angegeben wird. Dieses ist als Verhältnis einer Masse zum Massenunterschied der nächsten noch getrennt erscheinenden Masse (R = m / dm) definiert. Zum Beispiel würde man bei einem Auflösungsvermögen von 4000 die Peaks bei 4000 Th und 4001 Th noch getrennt sehen, aber ebenso die Peaks bei 2000 Th und 2000,5 Th da 2000/(2000,5 − 2000) = 4000. In der Praxis werden die beiden Begriffe Auflösung und Auflösungsvermögen leider oft nicht exakt auseinander gehalten.
Es gibt drei verschiedene Definitionen der Auflösung:
- Bei der 10-%-Methode definiert man dm als die Massenabweichung, bei der die Intensität eines Peaks auf 10 % des Maximums absinkt.
- Bei der 50-%-Methode definiert man dm als die Massenabweichung, bei der die Intensität eines Peaks auf 50 % des Maximums absinkt.
- Bei der Halbwertsbreitenmethode (FWHM (full width at half maximum height)) definiert man dm als die volle Peakbreite bei der halben Peakhöhe.
Die Massengenauigkeit gibt an, wie genau die Masse des Teilchens bestimmt werden kann. Diese Angabe erfolgt oft in parts per million (ppm), d. h., ein Molekül mit der nominellen Masse 500 kann bei einer Genauigkeit von 1 ppm auf 0,0005 u genau bestimmt werden.
Aufbau eines Massenspektrometers
 Schematische Zeichnung eines hochauflösenden Sektorfeld-Massenspektrometers
Schematische Zeichnung eines hochauflösenden Sektorfeld-MassenspektrometersEin Massenspektrometer (MS) besteht aus einer Ionenquelle, einem Analysator und einem Detektor. Jedes dieser Bauteile existiert in verschiedenen Bauformen und Funktionsprinzipien, die prinzipiell frei kombinierbar sind, obschon bevorzugte Kombinationen existieren. Diese werden im Folgenden beschrieben.
Ionenquelle
In der Ionenquelle wird der Analyt ionisiert. Dies kann mit Hilfe verschiedener Methoden erfolgen. Die Wahl der Methode ist hauptsächlich abhängig von der Art der zu analysierenden Substanz und davon, wie schonend ionisiert werden soll. Die Ionen werden meistens mit einem elektrischen Feld aus der Ionenquelle extrahiert und in den Analysator übergeben. Die einzelnen Methoden werden im Abschnitt Ionisationsmethoden behandelt.
Analysator
Im Analysator oder Massenselektor werden die Ionen nach ihrer Masse (genauer: Masse-zu-Ladung-Verhältnis, also m/q) getrennt. Es gibt mehrere sehr unterschiedliche Methoden, wie diese Massentrennung erfolgt. Abhängig von der Methode ist auch das Trennvermögen recht unterschiedlich. Die einzelnen Trennmethoden werden im Abschnitt Arten von Analysatoren behandelt.
Detektor
Der Detektor dient zur Erfassung der zuvor separierten Ionen. Als Detektor eingesetzt werden können Photomultiplier, Sekundärelektronenvervielfacher (SEV), Faraday-Auffänger, Daly-Detektoren, Mikrokanalplatten( MCP) oder Channeltrons. Der SEV wird teilweise in Kombination mit einer Konversionsdynode verwendet, bei der die Ionen aufgrund einer angelegten hohen Beschleunigungsspannung (bis zu 25 kV) auf eine Metalloberfläche prallen und der SEV dann die freiwerdenden Elektronen detektiert. In der Anfangszeit der Massenspektrometrie wurden auch Fotoplatten benutzt.
FT-ICR- und Orbitrap-Massenspektrometer messen Ströme (engl. image-currents), welche durch die sich bewegenden Ionenpakete in den Detektorplatten erzeugt werden. In diesem Fall werden die Ionen also nicht vom Detektor absorbiert und können deshalb mehrfach gemessen werden. Das trägt entscheidend zur hohen Messgenauigkeit dieser Instrumente bei.
Ionisationsmethoden
Elektronenstoßmethoden
- Elektronenstoßionisation (EI, auch Elektronenstoß, engl. electron impact)
- Die Elektronenstoßionisation ist die meistgenutzte Ionisationsmethode. [16] Dabei werden von einem Filament Elektronen mittels eines elektrischen Feldes auf eine kinetische Energie von 5 bis 200 eV (aus Stabilitätsgründen verwendet man oft 70 eV; außerdem ist der Ionisationsquerschnitt bei dieser Energie für die meisten Gase maximal) beschleunigt und durch eine Gaswolke der zu ionisierenden Moleküle geschickt. Beim Zusammenstoß der Elektronen mit den Molekülen wird ein weiteres Elektron herausgeschlagen, es entsteht ein Radikalkation. Dieses ist meistens instabil und zerfällt in kleinere Massenfragmente, von denen eines geladen bleibt. Die Größen und Häufigkeiten der Fragmente sind bei gegebener Beschleunigungsspannung der Elektronen für eine Substanz in Datenbanken katalogisiert und können zur Identifizierung herangezogen werden.
- Chemische Ionisation (CI)
- Bei der chemischen Ionisation wird ein Gas zugeführt, das durch Elektronenstoßionisation angeregt/ionisiert wird. Die aus dem Gas gebildeten Ionen reagieren dann mit dem Analyten und ionisieren ihn. Der Fragmentierungsgrad ist geringer als bei der Elektronenionisation.
- Sanfte Ionisation
- Bei der sanften Ionisation werden Gase mit geringer Ionisationsenergie von 10 bis 14 eV zur Ionisation der Probengase verwendet (z. B. Xenon, Argon oder Quecksilber). Dazu wird eine Ionenwolke des Ionisationsgases mittels Elektronenbeschusses erzeugt. Durch eine Auswahl über Vorfelder erreichen nur einfach ionisierte Atome des Ionisationngases das zu ionisierende Probengas. Eine Fragmentierung der Moleküle des Probengases wird dadurch nahezu ganz unterbunden. Da sich zudem unterschiedliche Gase mit gleicher Massenzahl von den Ionisierungsgasen verschieden ionisieren lassen, ist eine Differenzierung von Isomeren durch analytische Vergleiche möglich.
Feldionisationsmethoden
- Feldionisation (FI)
- Bei der Feldionisation wird ein Gas in einem starken elektrischen Feld an zahlreichen Spitzen (Graphitdendriten) sehr schonend ionisiert.
- Felddesorption (FD)
- Bei der Felddesorption wird ein fester oder flüssiger Analyt, der den zahlreichen Graphitdendriten in Lösung zugeführt wird, in einem hohen elektrischen Feld wie bei FI sehr schonend ionisiert.
- Liquid Injection Field Desorption Ionization (LIFDI)
- Bei der LIFDI wird durch den Druckunterschied zwischen Normaldruck und Vakuum ein flüssiger Analyt über eine Kapillare direkt auf den Graphitdendriten gespritzt und dort in einem hohen elektronischen Feld wie bei FI sehr schonend ionisiert. Da die Zuführung des Analyt durch eine Kapillare erfolgt, ist diese Methode speziell für das Analysieren von luftempfindlichen Substanzen geeignet.
Teilchenbeschussmethoden
Flüssigkeiten und Feststoffe können mit schnellen Atomen oder Ionen beschossen werden, worauf sich Ionen lösen. Kommen Atome zum Einsatz, heißt die Methode FAB (engl. Fast Atom Bombardment, Schneller Atombeschuss), bei Ionen SIMS (engl. secondary ion mass spectrometry, Sekundärionen-Massenspektrometrie). Neben den Sekundärionen werden auch ungeladene Teilchen (Sekundärneutralteilchen) erzeugt. Wenn diese zum Beispiel mit Laserlicht nachionisiert und dann analysiert werden, spricht man von Sekundär-Neutralteilchen-Massenspektrometrie (SNMS).
Sprühmethoden
- Elektrospray-Ionisation
- Bei der Elektrospray-Ionisation (ESI) werden Lösungen geladener oder polarer Substanzen versprüht, ionisiert und die Tröpfchen dann getrocknet, so dass Ionen des Analyten zurückbleiben. Diese Methode ist besonders geeignet für größere Moleküle, wie beispielsweise Proteine.
- Atmospheric Pressure Chemical Ionization
- Die chemische Ionisation unter Atmosphärendruck (engl. Atmospheric Pressure Chemical Ionization, APCI) funktioniert ähnlich wie ESI, nur dass die Lösung des Analyten vor der Ionisation verdampft wird. Die Lösemittelmoleküle werden an einer spitzen Elektrode bei Atmosphärendruck ionisiert. Die Methode ist auch für weniger polare Analyten geeignet.
Photoionisationsmethoden
- Ein-Photonen-Ionisation
- Bei der Ein-Photonen-Ionisation (engl. Single Photon Ionization, SPI) wird die Ionisationsenergie durch ein einzelnes Photon überwunden, dessen Wellenlänge im Vakuum-Ultraviolett-Bereich (VUV) liegt. Diese hochenergetischen Photonen können entweder als Synchrotronstrahlung oder mit Entladungslampen erzeugt werden. Ein weit verbreiteter Spezialfall ist die Atmosphärendruck-Photoionisation (engl. Atmospheric Pressure Photo Ionization, APPI).
- Resonanzverstärkte Mehrphotonenionisation
- Die resonanzverstärkte Mehrphotonenionisation (engl. Resonance Enhanced Multi Photon Ionization, REMPI) basiert auf der nahezu gleichzeitigen Absorption mehrerer ultravioletter Photonen die mittels eines Lasers erzeugt werden, deren Gesamtenergie oberhalb der Ionisationsenergie des zu ionisierenden Moleküls liegt. Auch zu dieser Ionisationsmethode existiert eine Athmosphärendruckvariante die Atmosphärendruck-Laserionisation (engl. Atmospheric Pressure Laser Ionization, APLI).
Die beiden Photoionisationsmethoden unter Atmosphärendruck werden hauptsächlich bei der Kopplung von Massenspektrometern mit LC-Systemen verwendet. Der Eluent wird zunächst verdampft und anschließend durch Photonen ionisiert. Dabei werden die Photonen senkrecht zum Molekülstrahl ausgesandt.
- Matrix-unterstützte Laser-Desorption/Ionisation
- Bei der Matrix-unterstützten Laser-Desorption/Ionisation (MALDI) wird der Analyt mit einem großen Überschuss einer Matrix genannten Substanz gemischt und cokristallisiert (Einbindung des Analyten in die kristalline Matrix). Die Matrix hat die Eigenschaft, beim Beschuss mit einem Laser bestimmter Wellenlänge Energie zu absorbieren (zum Beispiel Stickstofflaser 337 nm). Durch den Beschuss mit dem Laser wird der intakte Analyt verdampft, an den ein von der Matrix bereitgestellter Ladungsträger, z. B. ein Proton, gebunden ist.
Weitere Methoden
- Thermische Ionisation (TIMS, thermische Ionisationsmassenspektrometrie) wird in der Festkörpermassenspektrometrie eingesetzt. Dabei wird die Probe (Probenmenge je nach Stoff ng bis µg) zum Beispiel auf ein Wolframfilament aufgebracht. Durch das Filament wird Strom geschickt, wobei es sich erhitzt und die aufgebrachte Probe verdampft. Ein Teil der abgedampften Atome wird dabei ionisiert.
- Ein induktiv gekoppeltes Plasma (engl. inductively coupled plasma, ICP) bricht die meisten Verbindungen in ihre Elemente auf und es entstehen vorwiegend einfach positiv geladene Ionen. Diese Methode wird daher vor allem bei der Massenspektrometrie mit induktiv gekoppeltem Plasma in der anorganischen Elementanalytik und Spurenanalytik eingesetzt.
- Bei der Ionisation durch Glimmentladung (engl. glow discharge ionization, GDI) wird die Probe durch ein durch einen Lichtbogen erzeugtes Plasma ionisiert.
Arten von Analysatoren
Massenspektrometer werden durch den jeweilig eingesetzten Analysator typisiert.
Sektorfeld-Massenspektrometer
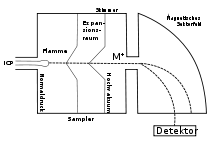 Schematische Zeichnung eines ICP/Sektorfeld-Massenspektrometers
Schematische Zeichnung eines ICP/Sektorfeld-MassenspektrometersIn Sektorfeld-Massenspektrometern werden die Ionen in elektrischen und magnetischen Feldern abgelenkt. Der Radius der Kreisbahnen, die sie in den Feldern durchlaufen, hängt von der Energie (im elektrischen Feld) und vom Impuls (im magnetischen Feld) der Ionen ab. In Kenntnis der Ladung, der Energie und des Impulses kann dann die Masse bestimmt werden. Sektorfeld-Massenspektrometer können so gebaut werden, dass Ionen mit leicht unterschiedlicher Geschwindigkeit auf einem Punkt im Detektor abgebildet werden (Geschwindigkeitsfokussierung). Auch Ionen, deren Flugbahn leicht geneigt ist, können auf einen Punkt abgebildet werden (Richtungsfokussierung). Massenspektrometer, die beides gleichzeitig können, nennt man doppelfokussierend. Die Fokussierung ist nötig, um bei hoher Auflösung noch eine akzeptable Intensität des Messsignals zu erhalten. Sektorfeld-Massenspektrometer erreichen Auflösungen von bis zu 100.000 und waren vor der Entwicklung der FT-Ionenfallen die Massenspektrometer mit der größten Auflösung. Heute werden sie nur noch selten verwendet, zum Beispiel in der Stabilisotopenmassenspektrometrie.
 Massenspektrometer zur Bestimmung von 16O/18O und 12C/13C Isotopenverhältnissen an biogenem Karbonat
Massenspektrometer zur Bestimmung von 16O/18O und 12C/13C Isotopenverhältnissen an biogenem KarbonatQuadrupol-Massenspektrometer
Im Quadrupol-Massenspektrometer werden die erzeugten Ionen durch ein statisches, elektrisches Feld beschleunigt und durchfliegen zentral vier parallel liegende Stabelektroden, deren Schnittpunkte mit einer Ebene senkrecht zur Zylinderachse ein Quadrat bilden (Quadrupol). Im Wechselfeld zwischen den Quadrupol-Stäben findet eine m/q-Selektierung statt, so dass jeweils nur Teilchen mit einer definierten Masse das Feld durchlaufen können. Die Ionen treffen in einem Detektor mit Messverstärker auf, der den Ionenstrom misst und von der Software des angeschlossenen PCs zur Zählrate bzw. zum Partialdruck umgerechnet wird. Die Ionisationseinheiten, Quadrupole und Detektoren sind in unterschiedlichsten Varianten für unterschiedliche Anwendungen erhältlich. Es ist in teuren, hochauflösenden aber auch günstigen Varianten (als Restgasanalysator) zu erhalten und hat dadurch eine hohe Verbreitung im F&E-Bereich.
 Triple-Quadrupol-Massenspektrometer
Triple-Quadrupol-Massenspektrometer Quadrupol eines Triple-Quadrupol-Massenspektrometer
Quadrupol eines Triple-Quadrupol-MassenspektrometerMassen-Selektierung im Quadrupol-Feld
Die gegenüberliegenden Elektroden des Quadrupol befinden sich auf gleichem Potential und zwischen benachbarten Elektroden wird eine Spannung mit einem Gleich- und einem hochfrequenten Wechselspannungs-Anteil angelegt, d. h.
 . Die Bahn der Ionen im QMS wird durch die Differentialgleichungen von Mathieu beschrieben. Aus systematischen Untersuchungen dieser Differentialgleichungen ist bekannt, dass es gewisse stabile und instabile Bereiche gibt. Die Arbeitsgerade, d. h., die Gerade, auf der alle beobachtbaren Massen liegen, wird durch das Verhältnis
. Die Bahn der Ionen im QMS wird durch die Differentialgleichungen von Mathieu beschrieben. Aus systematischen Untersuchungen dieser Differentialgleichungen ist bekannt, dass es gewisse stabile und instabile Bereiche gibt. Die Arbeitsgerade, d. h., die Gerade, auf der alle beobachtbaren Massen liegen, wird durch das Verhältnis  bestimmt. Um ein möglichst gutes Auflösungsvermögen (in der Praxis R = 1.000 bis 4.000) zu erreichen, muss
bestimmt. Um ein möglichst gutes Auflösungsvermögen (in der Praxis R = 1.000 bis 4.000) zu erreichen, muss  gelten. Der Schnitt aus Arbeitsgeraden und stabilem Bereich der Mathieuschen Differentialgleichungen ist dann sehr klein. Der Wert 0,1678 darf aber auf keinen Fall überschritten werden, ansonsten sind alle Ionen instabil. Instabile Ionen kollidieren während des Durchlaufes mit einem der vier Stäbe.
gelten. Der Schnitt aus Arbeitsgeraden und stabilem Bereich der Mathieuschen Differentialgleichungen ist dann sehr klein. Der Wert 0,1678 darf aber auf keinen Fall überschritten werden, ansonsten sind alle Ionen instabil. Instabile Ionen kollidieren während des Durchlaufes mit einem der vier Stäbe.Über die Einstellung der Frequenz ω oder Spannungen U,V lässt sich festlegen, welche Teilchen mit welchem Masse-zu-Ladungs-Verhältnis den Detektor über die zentrale Flugbahn erreichen. Die Bahn eines Teilchens mit richtigem m/e-Verhältnis ist sinusförmig mit gleichbleibenden Abständen zur Mittelbahn des Quadrupols. Alle anderen Ionen fliegen zwar auch im Sinustakt durch das Wechselfeld um diesen Sollbahnbereich, werden aber immer weit herausbeschleunigt, so dass sie irgendwann seitlich außerhalb des Quadrupols hinausschießen und den Einflussbereich des EM-Feldes verlassen.
Da die Ionisierung durch Elektronenstoß erfolgt, treten bei fast allen Atomen/Molekülen statt einem Peak bei der entsprechenden Masse des Atomes/Moleküles mehrere Peaks im Massenspektrum auf. Dies hat verschiedene Ursachen:
- Die Moleküle können bei der Kollision mit den Elektronen in verschiedene Bestandteile zerbrechen (Fragmentierung). So ruft z. B. Ethanol (C2H5OH) (Masse 46,07 g/mol) einen schwachen Peak bei 46 Th (C2H5OH) hervor, einen stärkeren Peak bei 45 Th (ein Wasserstoffatom fehlt) und den stärksten Peak (ca. 10-mal höher als die anderen Peaks) bei 31 Th (CH2OH). Weitere Peaks sind u. a. 17 Th (OH).
- Eine teilweise Rekombination oder Neubildung von Molekülen ist zwar sehr viel seltener. Es ist aber wegen der hohen Genauigkeit des Quadrupol-Massenspektrometers nicht auszuschließen, dass entsprechende Zählraten auftreten können.
- Die Bruchstücke der Moleküle bzw. Atome können zweifach oder sogar dreifach ionisiert werden. Da das Quadrupol-Massenspektrometer – wie andere Massenspektrometer – prinzipbedingt nur den m/q-Wert misst, ergeben sich folglich mehrere Peaks, z. B. Argon bei 40 Th und bei 20 Th.
- Bei hohen Konzentrationen von Edelgasen in einer Probe sind die sogenannten Cluster zu sehen, z. B. für Argon 40 ein Peak bei 80AMU.
Flugzeitmassenspektrometer (TOFMS)
Im Flugzeitmassenspektrometer (TOFMS) wird ausgenutzt, dass die Ionen beim Eintritt in den Analysator alle die gleiche Energie haben und leichte Ionen deshalb schneller sind als schwere. Daher erreichen beim Flug durch einen feldfreien Raum leichte Ionen den Detektor eher als schwere Ionen. Der Flugzeitanalysator besteht somit nur aus einem Rohr unter Vakuum mit einem sehr schnellen Detektor am Ende. Die Auflösung beträgt bis zu R = 15.000 (10-%-Methode). In der Praxis haben sich Geräte mit Ionenspiegeln bzw. Reflektron bewährt, bei denen die Flugstrecke durch ein zusätzliches elektrisches Feld am Ende der ursprünglichen Flugrichtung verdoppelt wird. Zusätzlich erreicht man durch diese Technik eine weitere Fokussierung, die die Varianz in der Geschwindigkeit der Ionen aufgrund des Doppler-Effekts minimiert.
Ionenfallen-Massenspektrometer
 Ionenfallen-Massenspekrometer
Ionenfallen-MassenspekrometerIn Ionenfallen-Massenspektrometern werden die Ionen durch elektromagnetische Felder in einem definierten Bereich gehalten und können so analysiert und manipuliert werden. In Ionenfallen-Massenspektrometern ist eine mehrfache Wiederholung von Anregung und Massenselektion möglich, ohne dass ein weiteres Bauteil benötigt wird. Folgende Typen von Ionenfallen-Massenspektrometer existieren:
- Quadrupol-Ionenfalle
- Linear trap
- Fouriertransformations-Ionenzyklotronresonanz (FT-ICR)
- Orbitrap
MS/MS (auch Tandem-Massenspektrometrie)
Um Fragmentierungen zu studieren oder auch um die Selektivität und Sensitivität (Nachweisgrenze) einer Quantifizierungsmethode entscheidend zu verbessern, koppelt man entweder mehrere Analysatoren hintereinander oder arbeitet in Ionenfallen. Zwischen zwei Analysatoren wird eine sogenannte Kollisionszelle eingebaut, um den Ionen durch Stöße mit einem Inertgas (meist Stickstoff oder Argon) Energie zuzuführen. Daraufhin zerfallen die Ionen sehr spezifisch zu anderen (leichteren) Ionen.
Viele Kombinationen der Analysatoren sind denkbar. Die gängigsten sind Triple-Quadrupol (QqQ), Doppel-Quadrupol (Qq), Tandem-TOF (TOF-TOF) und inzwischen auch TRAP-FTICR und TRAP-Orbitrap.
Am weitesten verbreitet sind sogenannte Triple-Quadrupol (QqQ, auch triple quads genannt). Dabei wird meist durch ESI ein Pseudomolekülion produziert, im ersten Analysatorquadrupol isoliert und dann im zweiten Quadrupol – der sogenannten Kollisionszelle oder Stoßkammer – angeregt.
In die Stoßkammer kann ein Stoßgas (meist Argon, Helium oder Stickstoff) eingespeist werden. Dabei wird der Druck so gewählt, dass im Mittel ein erzeugtes Ion maximal einmal mit einem Gasmolekül kollidiert. Diese Methode ermöglicht es, erzeugte Ionen weiter zu fragmentieren.
Der dritte Quadrupol gibt die Möglichkeit zu „scannen“, also alle Produktionen des im ersten Quadrupol isolierten Ions (engl. parent ion) zu ermitteln, oder selektiv nur ein bekanntes Fragmention zu beobachten. Durch das Erfassen aller Fragmentionen können Rückschlüsse auf die Struktur gezogen werden. Durch Beobachtung von nur ein oder zwei Fragmentionen, kann sehr empfindlich und selektiv quantifiziert werden. Diese Technik wird auch als Multiple Reaction Monitoring (MRM) bezeichnet.
Daneben gibt es auch andere Techniken für MS/MS (und sog. MSn). In Ionenfallen kann man ein Ion isolieren, und ihm dann entweder durch Kollision (hier aber meist mit Helium) oder auch durch Strahlung Energie zuführen. Gerade in FT-ICR-Geräten verwendet man dazu Infrarotlaser oder „Elektronenkanonen“ (engl. Electron Capture Dissociation). Eine weitere Methode ist die Electron Transfer Dissociation (ETD), die gerade im Bereich der Proteinidentifikation erste Einsatzmöglichkeiten erfährt.
Kopplung mit Chromatographieverfahren
Bei sehr komplexen Proben ist es nützlich, diese mit einem vorgelegten Trennverfahren aufzutrennen, bevor man sie dem Massenspektrometer zuführt. In diesem Sinn wird Massenspektrometrie oft zusammen mit Gaschromatographie (GC/MS) oder Flüssigchromatographie (LC/MS) betrieben. Weniger weit verbreitet sind die Kopplung mit Kapillarelektrophorese (CE/MS) und Ionenmobilitäts-Spektrometrie (IMS/MS). Teilweise werden auch mehrdimensionale Trenntechniken eingesetzt z. B. GCxGC-MS. Flugzeitmassenspektrometer eignet sich besonders gut im Verbund mit mehrdimensionaler Gaschromatographie, weil damit sehr schnell Massenspektren über einen großen m/q-Bereich aufgenommen werden können. Dieses Verfahren erlaubt eine genaue Auftrennung und Detektion verschiedener Verbindungsklassen aus komplexen Matrices (z. B. Erdölproben). Dafür werden zwei GC-Säulen mit unterschiedlicher Polarität hintereinander geschaltet.
Auswertung der Massenspektren
Voraussetzung für die Bestimmung der Masse m ist die Kenntnis der Ladung q des Ions, denn die Analysatoren können die Ionen nur nach dem Verhältnis m/q trennen. q ist jedoch immer ein ganzzahliges Vielfaches der Elementarladung e: q = z·e, und meistens ist z = +1 (einfach positiv geladen). Als Einheit von m/q wurde das Thomson Th vorgeschlagen: [m/q] = Th.
Zu beachten ist, dass es sich bei Massenspektren um Histogramme handelt, nicht um stetige Kurven.
Zunächst muss die Masse des Analyten bestimmt werden. Normalerweise ist das die Masse des schwersten detektierten Ions (Molekülpeak oder Molekülion). Allerdings ist bei der Elektronen-Ionisation oft ein Großteil der Moleküle gespalten. Testweise kann die Elektronenenergie verringert werden, so dass weniger Moleküle gespalten werden und der Molekülpeak deutlicher sichtbar wird.
Die weitere Auswertung basiert darauf, dass die Atome der verschiedenen chemischen Elemente einen unterschiedlichen Massendefekt haben. Daher kann aus einer sehr exakt bestimmten Masse eine Liste möglicher Summenformeln angegeben werden. Bei leichten Molekülen gibt es nur eine oder wenige passende Elementarzusammensetzungen. Mit steigender Masse oder zunehmender Anzahl an Heteroatomen steigt auch die Anzahl möglicher Kombinationen stark an.
Bei schwereren Molekülen stehen deshalb sehr viele mögliche Summenformeln zur Auswahl. Weitere Hinweise liefern die Isotopenzusammensetzungen der verschiedenen Elemente. So besteht der Kohlenstoff zum Beispiel zu 98,9 % aus 12C und zu 1,1 % aus 13C. Je nachdem, wie viele C-Atome im Molekül vorhanden sind, sind neben dem Hauptsignal im Spektrum Nebensignale zu finden, die vom Hauptpeak um 1 Th, 2 Th etc. entfernt sind und ein charakteristisches Intensitätsverhältnis zum Hauptsignal haben. Halogene wie Chlor und Brom haben ebenfalls charakteristische Isotopenverhältnisse, die zur Identifizierung benutzt werden können.
Die genannten Methoden sind auch auf die Bruchstücke anwendbar. Moleküle brechen oft an charakteristischen Stellen. Aus der Masse der Bruchstücke und evtl. weiteren Informationen kann schließlich die Strukturformel bestimmt werden.
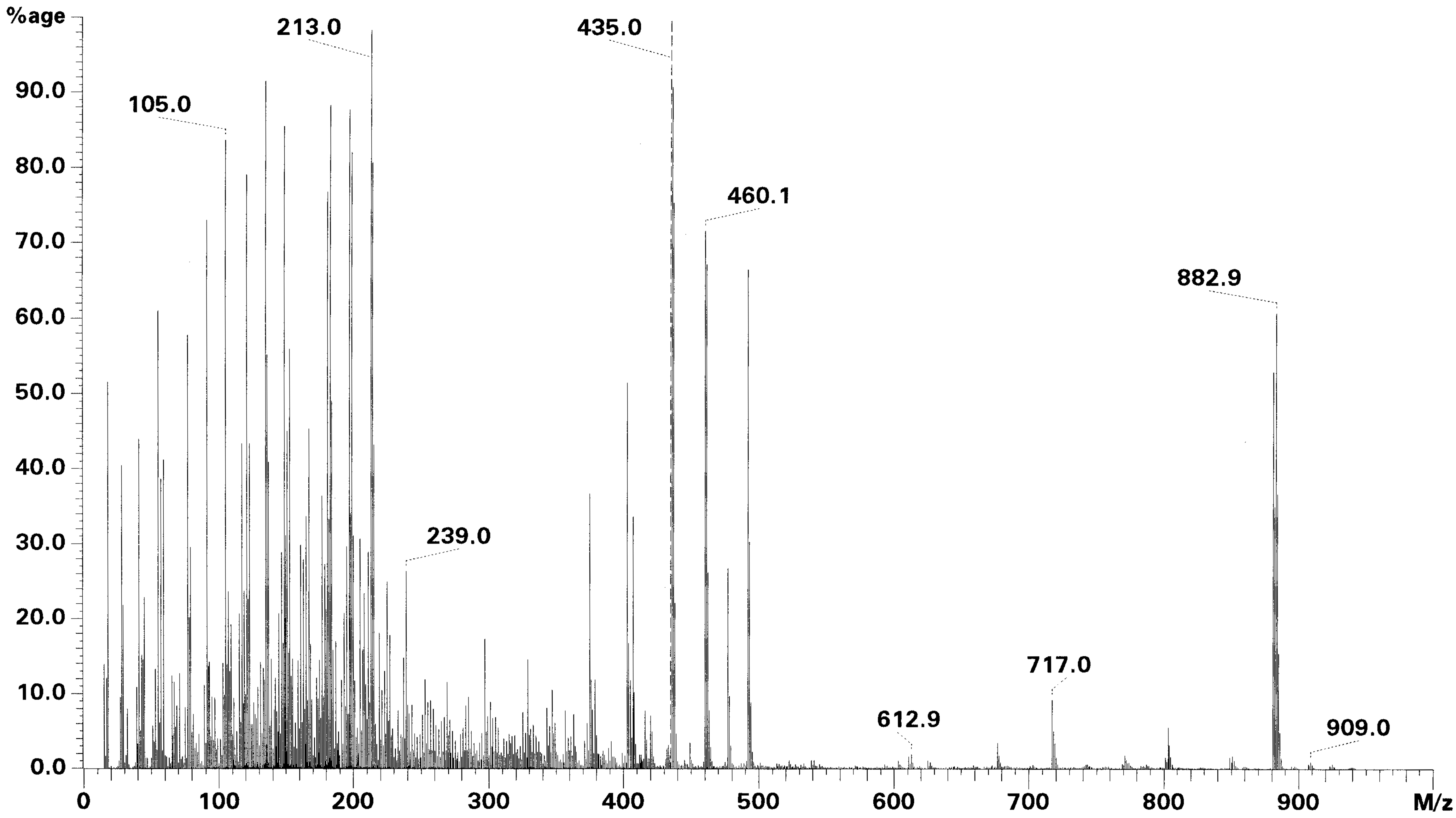
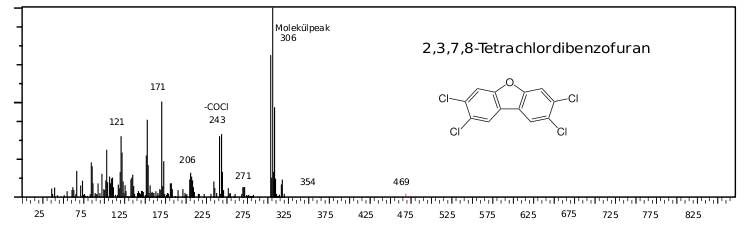 Beispiel eines Massenspektrums: Tetrachlordibenzofuran EI-positiv ionisiert
Beispiel eines Massenspektrums: Tetrachlordibenzofuran EI-positiv ionisiertDabei helfen vor allem bei mit positiver EI-Ionisierung erstellten Massenspektren auch Massenspektrenbibliotheken. Die bekanntesten sind unter den Kürzeln ihrer Vertreiber die Wiley- und die NIST-Massenspektrenbibliotheken. Durch den Peak Counting Score kann eine Identifizierung erfolgen.
Die Quantifizierung von Verbindungen wird bei der Massenspektrometrie dadurch erleichtert, dass bei der Analytik isotopenmarkierte (13C-markierte oder deuterierte) interne Standards verwendet werden können.
Ein Problem bei der Datenauswertung stellen die proprietären Datenformate der einzelnen Gerätehersteller dar [17]. Die Daten werden in eigenen Binärdatenformaten vorgehalten. Meist werden vom jeweiligen Hersteller in die eigene Steuerungs- und Managementsoftware integrierte Auswertungsprogramme mitgeliefert. Um Programme von Dritten zu benutzen, bedarf es oft der Datenkonvertierung zum Datenexport, für die es im Bereich der Forschung frei erhältliche Lösungen gibt. [18]
Anwendungen
Die Massenspektrometrie dient in der Analytik bzw. der analytischen Chemie als Analyseverfahren zur Bestimmung chemischer Elemente oder Verbindungen. In dieser Form werden Massenspektrometer in vielen Bereichen der Naturwissenschaften und der Technik für die Analyse von Materialien eingesetzt, unter anderem in der Chemie, Biologie, Archäologie und Klimatologie.
Auch in der Teilchenphysik werden Massenspektrometer verwendet. In diesem Bereich ist das Ziel jedoch weniger die Analyse von chemischen Elementen, sondern die Ermittlung der Massen von Elementarteilchen oder Atomkernen sowie der Detektion von noch unbekannten Teilchen.
Chemie
Für einen Analyten (die zu testende Substanz) wird die Häufigkeit, mit der geladene Moleküle (Ionen) und deren Massenfragmente auftreten, bestimmt. Die Massenspektrometrie ist eine wichtige Methode der analytischen Chemie bei der Aufklärung der Struktur und Zusammensetzung von Verbindungen und Gemischen. Der qualitative (Erkennung von unbekannten Substanzen) und quantitative (wie viel Substanz einer Verbindung ist vorhanden) Nachweis sehr kleiner Substanzmengen (ca. > 10−15 g = 1 fg (Femtogramm)) ist möglich.
Archäologie
Isotopenverhältnisse einiger Elemente erlauben Rückschlüsse auf die Ernährung der Menschen, deren Knochen untersucht werden. Siehe auch Isotopenuntersuchung. Das Isotopenverhältnis von Kohlenstoff 14C zu 12C im organischen Material von archäologischen Funden erlaubt es, die Zeit seit der pflanzlichen Bildung der vermessenen Substanz zu ermitteln. Zur Messung von 14C wird die Beschleuniger-Massenspektrometrie herangezogen.
Biologie
Massenspektrometrie wird in der Proteomik und Metabolomik verwendet, wo die Verwendung weitgehend jener in der Chemie entspricht. Biologische Proben, insbesondere Proteine, verlangen jedoch aufgrund der Molekülgröße und der speziellen Fragestellung (Identität, Sequenz, chemische Modifikation) bei der Aufklärung von systemischen Zusammenhängen eine besondere Probenvorbereitung und Messmethodik.
Klimatologie
Das Verhältnis der Häufigkeit bestimmter Isotope in Proben von Sedimenten und Eisbohrkernen lässt Rückschlüsse auf das Klima der Vergangenheit zu. Zum Beispiel verdampft Wasser, das das Isotop 16O enthält, leichter als solches, das das Isotop 18O enthält. Eiszeiten, bei denen große Mengen des Wassers als Eisschild dem Wasserkreislauf entzogen werden, verschieben die Häufigkeit dieser Isotope im Meer und damit auch im neu fallenden Schnee. Aus der Sauerstoff-Isotopenstufe kann auf die Menge des Inlandeises zu der Zeit geschlossen werden, als die Probe gebildet wurde.
Technik
Die Massenspektroskopie wird auch in vielen technischen Bereichen genutzt. Sie kann beispielsweise bei der Endpunkterkennung von Ätzprozessen eingesetzt werden. Ein anderer Anwendungsbereich ist die Einstellung und Optimierung der Gaszufuhr bei Beschichtungsprozessen (genauer chemische Gasphasenabscheidung). Hierbei wird das Abgas nach der Reaktion hinsichtlich unverbrauchter Reaktionsgase untersucht und die Gaszufuhr entsprechend angepasst. Die Massenspektroskopie kann aber auch zur Analyse der abgeschiedenen Materialien eingesetzt werden. Dabei können mithilfe von SIMS auch Tiefenprofile erstellt werden, was unter anderem bei der Analyse von dünnen Schichten eingesetzt wird.
Literatur
Allgemein
- Herbert Budzikiewicz, Mathias Schäfer: Massenspektrometrie – Eine Einführung. Wiley-VCH, Weinheim 2005, ISBN 978-3-527-30822-4.
- Hans-Joachim Hübschmann: Handbook of GC/MS, Fundamentals and Applications. 2. rev. Ed. Wiley-VCH Verlagsgesellschaft mbH, Weinheim 2008, ISBN 978-3-527-31427-0.
- Fred W. McLafferty und Frantisek Turecek (Deutsche Übersetzung): Interpretation von Massenspektren, 4. Ed., Spektrum Akademischer Verlag GmbH, Heidelberg 1995, ISBN 0-935702-25-3.
- A. M. Boehm et al.: Command Line Tool for Calculating Theoretical MS Spectra for Given Sequences. Bioinformatics, 2004. 20(16): 2889-2891; doi:10.1093/bioinformatics/bth328.
- Alexander M. Lawson (Editor): Mass Spectrometry - Clinical Biochemistry - Principles/Methods/Applications. Walter de Gruyter & Co., Berlin/New York 1989, ISBN 3-11-007751-5.
Spektrensammlungen
Spektrensammlungen liegen sowohl in gedruckten Werken als auch in gerätekompatiblen Dateiformaten vor. Letztere werden heute meist bereits mit den Geräten angeboten und für die komfortable Auswertung von unbekannten Massenspektren eingesetzt.
- M. Spiteller, G. Spiteller: Massenspektrensammlung von Lösungsmitteln, Verunreinigungen, Säulenbelegmaterialien und einfachen aliphatischen Verbindungen, Springer Verlag Wien, New York (1973), ISBN 3-211-81117-6
- A. Cornu, R. Massot: Compilation of Mass Spectral Data, Index de Spectres de Masse, Vol. 1 & Vol. 2, 2. Ed., Heyden & Son Ltd. London, Philadelphia, Rheine, (1979) ISBN 0-85501-086-X
- K. Pfleger, H. Maurer, A. Weber: Mass Spectral and GC Data of Drugs, Poisons and Their Metabolites, Part I & II, VCH Verlagsgesellschaft mbH, Weinheim (1985), ISBN 3-527-26303-9
- NIST Standard Reference Database 1A, NIST/EPA/NIH Mass Spectral Library with Search Program: (Data Version: NIST 08, Software Version 2.0f) NIST-Spektrensammlung, auch unter Einschluss von Designerdrogen verfügbar.
Quellen
- W. Barger: Schulungsunterlagen zum ICP-MS Kurs 2006. LAS PerkinElmer (Germany) GmbH, Rodgau, unveröffentlicht.
- R. S. Houk, V. A. Fassel, G. D. Flesch, A. L. Gray, E. Taylor: Inductively Coupled Argon Plasma for Mass Spectrometric Determination of Trace Elements. In: Analytical Chemistry. 1980, 52, S. 2283.
- D. Skoog, J. Leary: Instrumentelle Analytik. Grundlagen, Geräte, Anwendung. Berlin 1996. (dt. Übersetzung der 4. Auflage von Principles of Instrumental Analysis, Orlando 1992).
- S. M. Nelms: ICP Mass Spectrometry Handbook. Oxford 2005.
- H. E. Taylor: Inductively Coupled Plasma-Mass Spectrometry. San Diego 2001.
- R. Thomas: Practica Guide to ICP-MS. New York, 2004.
Siehe auch
Weblinks
- Geschichte der Massenspektrometrie
- Massenspektrometrie – eine umfassende Darstellung der Theorie und der Technik
- Massenspektrometrie (en)
- Probenpräparation für Proteinidentifizierung mittels Nano-LC-MS
- MS-Anwendungsbeispiele in der Proteinanalytik
Einzelnachweise
- ↑ Eintrag: mass spectroscopy. In: IUPAC Compendium of Chemical Terminology (the “Gold Book”). doi:10.1351/goldbook.M03748 (Version: 2.2).
- ↑ Eintrag: mass spectrometry. In: IUPAC Compendium of Chemical Terminology (the “Gold Book”). doi:10.1351/goldbook.M03746 (Version: 2.2).
- ↑ William Prout: On the relation between the specific gravities of bodies in their gaseous state and the weights of their atoms. In: Annals of Philosophy. 6, 1816, S. 321–330 (Online).
- ↑ William Prout: Correction of a mistake in the essay on the relation between the specific gravities of bodies in their gaseous state and the weights of their atoms. In: Annals of Philosophy. 7, 1816, S. 111–113 (Online).
- ↑ E. Goldstein: Canalstrahlen. In: Sitzungsbericht der Preussischen Akademie der Wissenschaften. Band 691, 1886, S. 691–699.
- ↑ E. Rückardt: Zur Erinnerung an Wilhelm Wien bei der 25. Wiederkehr seines Todestages. In: Die Naturwissenschaften. 42. Jahrgang, Heft 3, 1955, S. 57–62 doi:10.1007/BF00589524.
- ↑ E. Rückhardt: Zur Entdeckung der Kanalstrahlen vor fünfzig Jahren. In: Die Naturwissenschaften. 24. Jahrgang, Heft 30, 1936, S. 465–467, doi:10.1007/BF01473963.
- ↑ J. J. Thomson: Cathode Rays. In: Philosophical Magazine. 44, 1897, S. 293, doi:10.1080/14786431003659214 (facsimile von Stephen Wright, Classical Scientific Papers, Physics, 1964).
- ↑ J. J. Thomson: Rays of positive electricity. In: Proceeding of the Royal Society A. 89, 1913, S. 1–20 (Digitalisat auf JSTOR, wie exzerpiert in Henry A. Boorse, Lloyd Motz: The World of the Atom. Vol 1, 1966 PDF).
- ↑ F. W. Aston: Kanalstrahlen und Atomphysik. In: Die Naturwissenschaften. 24. Jahrgang, Heft 30, 1936, S. 467–469, doi:10.1007/BF01473964.
- ↑ Gohlke, R. S., Time-of-flight mass spectrometry and gas-liquid partition chromatography. Anal. Chem. 1959, 31, 535-41
- ↑ Gohlke, R. S.; McLafferty, F. W., Early gas chromatography/mass spectrometry. J. Am. Soc. Mass Spectrom. 1993, 4, (5), 367-371.
- ↑ M.B. Comisarow and A.G. Marshall, Chem. Phys. Lett. 25, 282 (1974)
- ↑ Munson, M.S.B.; Field, F.H. J. Am. Chem. Soc. 1966, 88, 2621-2630. Chemical Ionization Mass Spectrometry. I. General Introduction.
- ↑ Beckey H.D. Field ionization mass spectrometry. Research/Development, 1969, 20(11), 26
- ↑ Teach/Me Instrumentelle Analytik. Abgerufen am 09. Dezember 2010.
- ↑ A. M. Boehm et al.: Extractor for ESI Quadrupole TOF Tandem MS Data Enabled for High Throughput Batch Processing. In: BMC Bioinformatics. 5. Jahrgang, 162, 2004, doi:10.1186/1471-2105-5-162.
- ↑ A. M. Böhm: Methoden zur effizienten Proteinidentifizierung anhand von Massenspektrometrie. Logos-Verlag, 2006









Wikimedia Foundation.